
Infecção pelo vírus Epstein-Barr e oncogênese
Epstein-Barr virus infection and oncogenesis
Antonio Vaz de Macedo1; Manoel Otávio da Costa Rocha2
1. Acadêmico do curso de Medicina da Faculdade de Medicina da Universidade Federal de Minas Gerais
2. Professor Titular do Departamento de Clínica Médica da Faculdade de Medicina da Universidade Federal de Minas Gerais
Prof. Manoel Otávio da Costa Rocha
Curso de Pós-Graduação em Medicina Tropical Faculdade de Medicina da UFMG
Av. Alfredo Balena, 130
Belo Horizonte - MG 30130-100
(31) 3248-9822
E-mail: rochamoc@medicina.ufmg.br
RESUMO
As infecções como um todo associam-se a 15% a 20% dos cânceres em geral. Entre os agentes etiológicos carcinogênicos, destacam-se os vírus oncogênicos. O vírus Epstein-Barr (VEB) é o mais potente vírus indutor de transformação e crescimento celular conhecido, sendo capaz de imortalizar linfócitos B humanos. Está relacionado com o linfoma de Burkitt, o carcinoma nasofaríngeo e outros tipos de neoplasia. Porém, não se sabe ao certo se o VEB seria apenas um componente inocente ou se contribui realmente para o desenvolvimento desses tumores. A compreensão da persistência do VEB no organismo e dos mecanismos pelos quais, na sua interação com a célula, ele contribui para o surgimento de uma neoplasia pode permitir novas abordagens para a prevenção e o tratamento dos tumores a ele associados.
Palavras-chave: Vírus oncogênicos, vírus Epstein-Barr, linfoma de Burkitt, neoplasias nasofaríngeas.
As infecções parecem constituir, depois do tabagismo, a mais importante causa prevenível de câncer em seres humanos. Entre 15% a 20% desses cânceres estão associados com as infecções (em torno de 7%, nos países desenvolvidos, e 22%, nos países em desenvolvimento), correspondendo a 1,2 milhão de casos por ano.1
Os vírus oncogênicos estão especialmente envolvidos nesse processo. O VEB está relacionado com o carcinoma nasofaríngeo (CNF), linfoma de Burkitt (LB) e outros tipos de neoplasia; o vírus do papiloma humano (VPH), com o câncer anogenital; os vírus das hepatites B e C, com o carcinoma hepatocelular; e o vírus T linfotrópico humano do tipo I (VTLH-1), com a leucemia/linfoma de células T do adulto. Cabe lembrar o papel direto ou indireto da infecção pelo vírus da imunodeficiência humana (VIH) no desenvolvimento de diversas neoplasias.
Em todos os tumores associados com infecção viral, observa-se longo período de latência entre a infecção e o aparecimento da neoplasia e baixa proporção de indivíduos infectados que desenvolvem tumor maligno. Os vírus oncogênicos parecem ser, portanto, necessários, mas não suficientes para a indução das neoplasias a eles associadas − as condições no momento da infecção inicial, além de outros fatores, são essenciais, e podem determinar o padrão da interação agente-hospedeiro.2,3
Os vírus oncogênicos são capazes de persistir no organismo, podendo infectar precursores de células malignas e continuar a expressar genes virais em células tumorais. Para que uma neoplasia se desenvolva, entretanto, a infecção viral tem de afetar as células pluripotentes de um tecido, já que células diferenciadas, de maneira geral, não podem se imortalizar.3 Tumores que se desenvolvem como resultado de agentes infecciosos são quase sempre monoclonais, o que indica que eles teriam sua origem em uma única célula maligna. É interessante observar que a remoção do agente infeccioso pode reverter o desenvolvimento tumoral.
É preciso ter cautela ao se associar um tipo de neoplasia e um dado agente viral. O vírus pode, por exemplo, ser ativado como conseqüência de um processo carcinogênico e, dessa forma, não ser o agente etiológico do mesmo. Além disso, pode constituir um mero marcador de uma infecção por outro agente, o verdadeiro agente causal. Por outro lado, associações podem passar despercebidas caso se usem marcadores sorológicos inadequados, ou se o vírus for removido do genoma hospedeiro durante a carcinogênese.
O estudo da carcinogênese viral ajuda a compreender a carcinogênese em geral, na medida em que se conhecem os mecanismos pelos quais os vírus, na sua interação com a célula hospedeira, contribuem para o surgimento de uma neoplasia. O LB, por exemplo, constitui modelo único para o entendimento das várias etapas envolvidas nesse processo. Nele, o VEB aparece como provável iniciador dos processos oncogênicos.
INFECÇÃO VIRAL E CARCINOGÊNESE
Os vírus necessitam, para se replicar, da maquinaria biossintética celular. Na sua interação com a célula, as seguintes situações podem ocorrer:2,4
infecção produtiva, com lise celular: ocorre multiplicação do vírus em células permissivas, com inibição da biossíntese celular e conseqüente lise da célula hospedeira;
a informação genética viral permanece em estado latente na célula (sob a forma de epissoma ou incorporada ao genoma celular), mas sujeita à reativação subseqüente para um novo ciclo produtivo;
Essas diversas formas de infecção podem ser concomitantes em um mesmo indivíduo, desde que, é claro, afetem diferentes células-alvo.4
A presença do genoma viral pode afetar o controle do ciclo de divisão e diferenciação da célula hospedeira. Mais especificamente, a integração ao genoma celular pode resultar em mutagênese insercional , ao provocar deleções, transposições, duplicações (nos genomas viral e celular), translocações cromossômicas, amplificações do ADN hospedeiro, inserção de genes virais transformantes ou de promotores e transativadores virais na intimidade de oncogenes celulares, etc. Esses rearranjos podem ocorrer no próprio sítio de integração, ou mesmo à distância, uma vez que a inserção viral pode desencadear "instabilidade genética", favorecendo o acúmulo de mutações.2
A carcinogênese viral é um processo em múltiplas etapas, que envolve:2
Nos cultivos de células transformadas por vírus, observam-se células com morfologia alterada e que, devido à perda da inibição por contato, se dispõem em várias camadas, formando colônias desorganizadas. Nessas colônias, as células com o genoma viral integrado expressam um ou mais genes virais que podem promover a sua imortalização (capacidade de se reproduzir indefinidamente). Notam-se modificações antigênicas no nível de membrana, e outros novos antígenos são expressos, codificados por genes virais e celulares. Passadas várias gerações, as células transformadas são capazes de induzir tumores, ao serem inoculadas, por exemplo, em camundongos recém-nascidos ou atímicos, desfavorecidos pela imaturidade ou deficiência imunológica.
Em relação aos mecanismos gerais de oncogênese viral, citam-se:3
A proliferação normal das células é regulada por genes promotores de crescimento, denominados proto-oncogenes , e genes que restringem a proliferação celular, conhecidos como genes supressores tumorais . Qualquer alteração produzida nesse equilíbrio por alguma modificação na atividade desses genes pode iniciar a cascata de eventos que conduz a uma progressiva transformação maligna.2 Quaisquer genes envolvidos na regulação da expressão gênica ou da replicação do material genético, na progressão através de fases específicas do ciclo celular ou em mecanismos de sinalização extra-celular podem, potencialmente, funcionar como genes supressores tumorais.5
Proteínas celulares que, normalmente, são supressoras da indução tumoral interagem com proteínas virais transformantes. Duas delas, a p53 e a Rb105 , parecem estar envolvidas em um mecanismo central de regulação em células de mamíferos. A interação com proteínas celulares entregaria aos vírus oncogênicos o controle dos sistemas transcricionais ou regulatórios das células, como os responsáveis pelo controle do ciclo celular, o que poderia ser importante, talvez, para otimizar a replicação viral.6,7
As mutações na p53 são as alterações mais freqüentes nos cânceres humanos. Esta proteína desempenha papel central na vigilância e manutenção da estabilidade genômica logo após o dano genético celular. As mutações na p53 determinam sua inabilidade para deter o ciclo celular na fase G1 e induzir apoptose (morte celular programada). Proliferação celular desordenada também resulta de mutação na proteína Rb105 , a qual exerce função importante no controle do ciclo celular, através da ativação indireta de genes, como o c-myc, envolvido na replicação do ADN e na proliferação celular.
Além dos fatores virais e celulares implicados na carcinogênese, o estado imunológico do hospedeiro e fatores hormonais, genéticos e ambientais podem também estar envolvidos.
ASPECTOS ONCOGENÉTICOS DA INFECÇÃO PELO VEB
O VEB é o mais potente vírus indutor de transformação e crescimento celular conhecido, sendo capaz de imortalizar linfócitos B humanos. Linhagens celulares podem ser facilmente obtidas, in vitro , a partir do sangue de indivíduos que estiveram expostos ao VEB, ou se linfócitos são cultivados na presença desse agente. Portanto, o genoma do VEB é necessário para a obtenção de culturas contínuas de células linfoblastóides humanas.4,8 A confirmação de que o agente transformante é o VEB foi feita através da detecção de antígenos virais, por imunofluorescência indireta ou por hibridização in situ de ADN celular com ADN purificado de VEB, dentro do núcleo das células transformadas.9
No caso da mononucleose infecciosa (MI), a célula que inicia essas linhagens é de caráter não-maligno, carreando o genoma viral, in vivo , sob a forma não-produtiva. Quando transferidas para condições laboratoriais, tais células sofrem uma série de transformações, indistinguíveis, para alguns, das obtidas a partir de células de LB; estas, entretanto, já apresentam transformação maligna in vivo .
O VEB tem 10 genes transformantes (o genoma completo contém mais de 100 genes), um dos quais codifica uma proteína de membrana essencial para a imortalização, a PLM (proteína latente de membrana), cuja expressão só ocorre em células transformadas; por sua estrutura, ela poderia ser um receptor para fatores de crescimento. A PLM-1 ativa fatores de transcrição e interage com moléculas celulares de sinalização, podendo ter papel importante na oncogênese associada ao VEB. É capaz de transformar morfologicamente linfócitos B e células epiteliais, além de prevenir a apoptose mediada pela p53 .10,11,12 Induz a expressão de bcl-2 , oncogene que promove a sobrevivência celular, e numerosas outras alterações fenotípicas.11 Não parece ser regularmente expressa na infecção lítica in vivo de linfócitos B pelo VEB. Durante a infecção latente in vitro de linfócitos B, são expressos os antígenos nucleares do VEB (ANVEB ), PLMs e pequenos ARNs do VEB (AVEB ). O ANVEB desempenha papel importante na imortalização dos linfócitos B; os AVEBs , por sua vez, constituem marcadores sensíveis da infecção latente pelo vírus.
A detecção de ADN, ARN e proteínas do VEB, bem como de respostas imunes vírus-específicas, tem permitido a associação do vírus com diversas afecções malignas. A reação em cadeia da polimerase (RCP) tem possibilitado a detecção precoce do ADN viral no líquor de alguns pacientes com SIDA e linfoma do sistema nervoso central (SNC), associado ao VEB. Além disso, por meio dela, é possível monitorar a quantidade de ADN viral no sangue de pacientes com doença linfoproliferativa. O envolvimento do VEB em tumores que não os do cérebro pode ser verificado pela demonstração in situ do ADN do vírus ou pela detecção de antígenos virais por técnicas imunocitoquímicas.
Cerca de 75% de todas as leucemias linfóides e 90% de todos os linfomas têm origem em linfócitos B. As leucemias linfocíticas agudas (LLA) são predominantemente cânceres de crianças e adultos jovens. A leucemia de Burkitt (de linfócitos B) ou do tipo L3, que constitui a leucemia linfóide clinicamente mais agressiva conhecida, ocorre em crianças de países em desenvolvimento, e parece estar associada à infecção pelo VEB. Responde por pequena parcela das LLAs na infância e na idade adulta.
Os linfomas de linfócitos T são mais freqüentemente associados com o VEB que os linfomas de linfócitos B. Isso sugere que os linfócitos T não sejam tão bem adaptados quanto estes à infecção por esse vírus e, portanto, sejam incapazes de sustentar uma infecção latente não-transformante.13
Em pacientes com tumores associados ao VEB, o estádio exato em que a infecção ocorre parece variar, podendo esta se dar antes da expansão clonal maligna (caso em que o VEB poderia desempenhar papel essencial no processo de carcinogênese) ou após a mesma, ou seja, em tumores já estabelecidos. Neste caso, a infecção poderia não ter efeito algum no processo ou, alternativamente, conferiria vantagem de crescimento aos subclones de células infectadas.13 Acredita-se que a célula infectada pelo VEB e em proliferação seria mais susceptível ao acúmulo de mutações genéticas, as quais, por sua vez, levariam à transformação maligna.
Fatores próprios do hospedeiro, como capacidade imunológica, condições genéticas e comportamentais e contato com outros agentes infecciosos, além de características intrínsecas do vírus, poderiam, talvez, explicar o fato de o VEB induzir proliferação maligna em apenas certas pessoas. A imunidade celular representa, provavelmente, parte importante do controle das neoplasias malignas. A compreensão da persistência do VEB no organismo e de seu papel oncogênico pode permitir novas abordagens para a prevenção e o tratamento dos tumores a ele associados.
LINFOMA DE BURKITT
Em 1958, Dennis Burkitt descreveu, pela primeira vez, um linfoma que incidia de maneira endêmica em certas regiões da África, acometendo, sobretudo, crianças.14 A partir do cultivo, in vitro , de células tumorais obtidas de biópsias de LB, Epstein et al. mostraram, em 1964, através da microscopia eletrônica, a presença de vírus, com morfologia típica dos herpesvírus, em pequena proporção dos linfócitos em cultura.15 Estudos virológicos e imunológicos subseqüentes demonstraram tratar-se de vírus diferente, que foi denominado "vírus de Epstein-Barr".16
Nas regiões africanas em que o LB é endêmico, a infecção primária pelo VEB ocorre, sobretudo, no grupo etário de um a três anos; entretanto, a incidência máxima desse linfoma encontra-se naquele de seis a oito anos.17 Isso indica que o desenvolvimento do tumor poderia estar relacionado a uma infecção primária tardia, rara na região, ou, o que é mais provável, a um evento secundário, que ocorreria alguns anos após a infecção primária.
Estudos demonstraram que os títulos médios geométricos dos anticorpos contra o antígeno do capsídeo viral (anti-ACV) do VEB eram oito a 10 vezes mais altos nos pacientes com LB que em controles normais.18,19 Mais de 85% dos pacientes com o linfoma tinham anticorpos contra os antígenos precoces (AP) do VEB - com notável predomínio do componente R e em títulos altos o suficiente para exceder, algumas vezes, os títulos de anti-ACV.20 Em controles normais, tais anticorpos, quando presentes, encontravam-se em títulos baixos. Títulos elevados de anti-AP-R parecem se correlacionar com maior probabilidade de recidiva da doença.19,20
Um estudo prospectivo iniciado na década de 70, em Uganda, do qual fizeram parte 42.000 crianças de um a cinco anos, revelou, até 1981, 14 casos de LB. Concluiuse que o risco de desenvolver esse linfoma naquela região africana era, aproximadamente, 30 vezes maior nas crianças com títulos de anti-ACV - duas ou mais diluições acima da média da população controle normal, o que corresponde a um risco superior àquele observado entre o tabagismo pesado e o carcinoma broncogênico.17,21
Acredita-se que existam duas formas distintas de LB: uma associada ao VEB, que ocorre principalmente em crianças, na África Central, onde é endêmico, e, esporadicamente, em outras partes do mundo; e uma outra, de ocorrência rara, não associada ao vírus, em todo o mundo, inclusive na África. Além disso, existe uma terceira forma, associada à imunodeficiência, que ocorre, por exemplo, em pacientes com SIDA.
Tem sido postulado que a causa do LB, nos países onde ocorre endemicamente, seria uma combinação da infecção pelo VEB e pelo Plasmodium falciparum . A malária pode deprimir o controle imunológico através da ativação de linfócitos T-supressores. Além disso, há evidências de que a malária aguda está associada com um aumento no número de linfócitos carreadores de VEB na circulação: esse vírus, agindo conjuntamente com o plasmódio, estimula a proliferação de linfócitos B infectados.22 O estado hiperproliferativo que daí sobrevém favoreceria o aparecimento de anormalidades citogenéticas nestas células, com conseqüente evolução para câncer.23
CARCINOMA NASOFARÍNGEO
A associação do VEB com o CNF (ou linfoepitelioma) surgiu com as observações de Old et al. de que o soro de pacientes com esta neoplasia comportava-se, em testes de dupla-difusão com extratos de culturas de linfócitos produtoras de VEB, de maneira idêntica ao soro de pacientes com LB.24,25,26,27 Demonstrou-se, mais tarde, que praticamente todos os pacientes com CNF tinham anticorpos contra o vírus, e que os títulos de anti-ACV eram bem maiores nestes pacientes que nos respectivos controles.28
O CNF, raro na América do Norte, é prevalente no Extremo Oriente (sobretudo entre chineses do Sudeste Asiático), em esquimós do Pólo Norte, no Alasca, no leste da África e em populações do norte deste continente, onde ocorre endemicamente. Nesses indivíduos, a infecção primária pelo VEB ocorre nos primeiros anos de vida, precedendo em muitos anos o aparecimento do tumor, o qual predomina em adultos jovens. O CNF pode, entretanto, acometer crianças, correspondendo a cerca de 1% das neoplasias nesta faixa etária. Tem sido também associado com fatores genéticos e ambientais, como o consumo de peixes salgados. Constitui a principal causa de morte entre adultos jovens do sul da China, sendo um dos cânceres mais comuns nessa região (corresponde a 20% de todos os cânceres em determinadas áreas29).
Em estudo realizado no Brasil, envolvendo três crianças com diagnóstico confirmado de CNF, foram observados, por métodos imuno-histoquímicos, antígenos PLM-1 do VEB no citoplasma de 50 a 60% das células tumorais. Destacou-se que, apesar da raridade dessa neoplasia na infância, é freqüente sua associação com a infecção pelo VEB.30 Entretanto, o papel exato desse vírus na patogênese do CNF ainda não está plenamente esclarecido. A expressão de PLM pode ser um importante fator causal deste tumor ou, se não essencial para a gênese do mesmo, pode ser vantajosa para a progressão tumoral.10 Poder-se-ia, ainda, atribuir um papel etiológico ao VEB baseando-se na constatação do epissoma clonal do vírus nas células tumorais, o que sugere uma possível expansão clonal a partir de uma célula progenitora infectada.31
Carcinomas não-diferenciados do tipo nasofaríngeo são raramente observados em outros sítios anatômicos. É provável que, na medida em que se analisarem números maiores de carcinomas, novas associações com o VEB sejam identificadas.13
DOENÇA DE HODGKIN
A doença de Hodgkin (DH) é mais freqüente em indivíduos de condição socioeconômica mais alta de populações ocidentais. Há dois picos de incidência da doença: um por volta dos 25 anos e outro após os 45 anos. Acredita-se que o VEB possa desempenhar papel direto ou indireto na patogênese de alguns casos de DH, notavelmente naqueles pacientes com títulos elevados de anticorpos para esse vírus (30% a 40% dos casos).32
Demonstrou-se, recentemente, a presença de ANVEB-1 e de ácidos nucléicos do VEB em tecido neoplásico obtido de 20% a 40% dos casos de DH, nos quais se observou uma proliferação oligo- ou monoclonal de linfócitos B infectados pelo vírus.12,33,34,35,36 Sabe-se que, além de ANVEB-1, PLM-1 e PLM-2 são expressos nesse tipo de tumor. Acredita-se que a expressão de PLM esteja relacionada a subtipos histológicos agressivos de DH.34 O ADN do vírus parece residir, primariamente, na célula de Reed-Sternberg e variantes, sendo encontrado, com maior freqüência, na população pediátrica, principalmente em tumores do tipo "celularidade mista".
A detecção do VEB na DH parece ser maior nos países em desenvolvimento, sobretudo nos latino-americanos, sendo a população infanto-juvenil de países como o Brasil possível alvo preferencial dos efeitos desse vírus.37 Os idosos, os infectados pelo VIH e os pacientes de países pobres apresentam, mais comumente, DH do tipo "celularidade mista" ou "depleção linfocitária". Porém, independentemente do tipo histológico, a presença do VEB não parece ter efeito significativo no prognóstico. Além disso, a variabilidade nos resultados de diversos estudos sugere que esse linfoma apresenta múltiplas etiologias. Quanto à MI, a ocorrência precoce dessa infecção poderia estar especificamente relacionada com o desenvolvimento da DH. Acredita-se que o risco de desenvolver esta doença seja duas a três vezes maior em indivíduos com história de MI do que o normalmente esperado.32
Em um estudo realizado no Ceará, com a finalidade de avaliar o perfil da DH infanto-juvenil nesse estado e sua possível associação com o VEB, demonstrou-se a presença do vírus em 29 (85,29%) dos 34 pacientes com confirmação histológica da doença. A PLM foi detectada em 27 dos 34 casos (79,41%), enquanto o AVEB o foi em 28 casos (82,35%). Em relação aos subtipos histológicos específicos, 100% dos casos de "celularidade mista" (cor-respondente a 64,70% do total de pacientes), 50% dos de "esclerose nodular" e o único caso de "depleção linfocitária" - além de um caso inclassificável - foram positivos para o VEB (não se diagnosticou o subtipo "predominância linfocitária" no estudo). Observou-se pico de incidência na faixa etária de 5 a 9 anos (47,05% dos casos). Os cinco casos negativos para o vírus foram todos do subtipo "esclerose nodular" e ocorreram em pacientes com mais de 15 anos. É importante destacar, entretanto, que o VEB constitui apenas um dentre vários co-fatores da DH infanto-juvenil brasileira: a heterogeneidade genética da população, somada a fatores ambientais, pode produzir diferentes padrões de resposta imunológica e susceptibilidades variadas ao vírus.37
INFECÇÃO PELO VEB E IMUNOSSUPRESSÃO
A MI constitui, via de regra, doença linfoproliferativa auto-limitada, já que a resposta imunitária celular permite o controle da ativação policlonal de linfócitos B. Entretanto, ocasionalmente, casos esporádicos de infecção primária pelo VEB evoluem para síndromes linfoproliferativas incontroladas.
Em todos os tipos de imunodeficiência, observa-se aumento na incidência de linfomas malignos e, embora haja certa heterogeneidade, na maioria dos casos, a característica marcante é a alteração da função citotóxica dos linfócitos T. A maioria dos linfomas, nesses casos, são do tipo LB ou de "células grandes difusas". Inicialmente, a proliferação de linfócitos B é claramente policlonal; logo, entretanto, parece haver uma seleção oligoclonal ou, ainda, monoclonal, que pode dar lugar ao aparecimento de células malignas.
Em transplantados, por exemplo, especialmente naqueles tratados com ciclosporina A, é relativamente comum o aparecimento de doenças linfoproliferativas associadas ao VEB. As desordens linfoproliferativas póstransplante ocorrem em 2,5% dos casos de transplantes renais, em 5% dos cardíacos e em 2% a 4% dos hepáticos.2 Quanto ao transplante alogênico de medula óssea, a prevalência é muito variável e depende de múltiplos fatores. Esses pacientes começam com uma infecção pelo VEB, a qual progride rapidamente para uma forma disseminada. Linfócitos B infectados proliferantes infiltram linfonodos e múltiplos órgãos, e os pacientes se apresentam com febre e linfadenopatia ou sintomas gastrointestinais. Estudos histopatológicos mostram hiperplasia de linfócitos B ou linfoma mono- ou policlonal.
Na imunossupressão induzida por medicamentos em pacientes transplantados, há um delicado balanço entre o tratamento necessário para evitar a rejeição do tecido transplantado ou a doença enxerto-versus -hospedeiro e as conseqüências danosas da reativação do VEB e de outros herpesvírus. Métodos de monitoração de pacientes, com a finalidade de predizer o aparecimento de linfomas, estão sendo investigados. Títulos decrescentes de anti-ANVEB-1 indicam controle inadequado sobre linfócitos B transformados pelo VEB. Isso pode estar relacionado com doença linfoproliferativa, mas esse achado é muito freqüente em indivíduos imunossuprimidos para que possa ser útil.38 O crescimento espontâneo de linfócitos B transformados e a presença de genomas do VEB no soro, medidos pela RCP, podem predizer o surgimento de linfomas;39 entretanto, são necessárias avaliações mais precisas para verificar a utilidade dessas observações.
INFECÇÃO PELO VEB, ONCOGÊNESE E SIDA
Nos pacientes imunodeprimidos, em especial naqueles com SIDA, as infecções provocadas pelos herpesvírus constituem causa importante de morbimortalidade. Essas enfermidades podem recorrer com mais freqüência e com curso mais grave e prolongado nessa condição. Devido à grande carga viral existente nestas situações, há, também, maior propensão para desenvolver resistência às drogas antivirais.40
Por constituírem infecções crônicas, pandêmicas, e serem mantidas sob controle pela imunidade celular, as infecções pelos herpesvírus tornaram-se mais prevalentes e graves após a eclosão da SIDA. Além disso, passaram a manifestar sintomatologia atípica quando associadas a esta doença.40 Um aspecto interessante dessas infecções, nessa condição, é a observação de Golden et al.41 de que antígenos de diversos herpesvírus, especificamente o vírus herpes simplex (VHS), o citomegalovírus (CMV) e o VEB, são capazes de estimular macrófagos a produzirem citocinas associadas ao aumento da expressão do VIH em células CD4+.
Na infecção pelo VIH, ocorrem linfomas extra-nodais agressivos, principalmente do SNC, devido à proliferação mono- ou policlonal de linfócitos B infectados pelo VEB. Genomas deste vírus estão presentes em cerca de um terço dos linfomas de linfócitos B e em virtualmente todos os casos de linfoma do SNC em pacientes com SIDA.40,42,43 O linfoma do SNC ocorre em cerca de 10% desses pacientes, sendo o tumor cerebral mais comum nesse grupo. A demonstração do VEB no líquor é forma diagnóstica importante, uma vez que genomas deste vírus podem ser aí detectados antes mesmo que os pacientes desenvolvam sintomas clínicos. Além disso, a presença do genoma viral no soro poderia constituir um marcador de linfomas generalizados associados ao vírus. Entretanto, o valor diagnóstico da RCP sérica está ainda sob investigação.
Para explicar o desenvolvimento do linfoma do SNC em pacientes imunossuprimidos, duas situações são possíveis:44
• os linfomas originar-se-iam de proliferações linfóides locais induzidas pelo VEB;
• o VEB seria conduzido, passivamente, ao SNC, dentro de linfócitos B atraídos por alguma infecção nessa região.
No Brasil, a incidência desse tipo de linfoma é desconhecida, e sua associação com o VEB tem sido pouco pesquisada. Para verificar esta associação, foram analisados 12 casos de linfoma primário do SNC, em pacientes brasileiros. Evidenciaram-se somente três casos positivos para o VEB, dois dos quais tinham história de imunossupressão associada a transplante renal e o outro, de SIDA. Nos demais casos, negativos para o VEB, não havia qualquer evidência clínica ou laboratorial de imunodeficiência. Concluiu-se que esse vírus parece ter papel importante no desenvolvimento do linfoma do SNC na presença de imunossupressão.44
O genoma do VEB foi também detectado em linfoma primário do SNC em pacientes sem imunodeficiência evidente. A detecção de ADN viral no tecido tumoral, mas não no tecido normal adjacente, indica provável indução do linfoma pelo vírus.45 Outras etiologias poderiam também estar implicadas.44
Em pacientes com SIDA, a infecção pelo VEB parece preceder a expansão clonal dos linfomas a ele associados. No entanto, a proliferação induzida pelo vírus é apenas uma das múltiplas etapas do processo de transformação neoplásica, estando também implicadas alterações genéticas, como a ativação de oncogenes (por exemplo, o c-myc ) e a inativação de genes supressores de tumor, como o p53 .43
A associação entre o VEB e tumores de músculo liso, em pacientes com SIDA e em transplantados, tem sido descrita, havendo relatos de leiomiossarcomas e leiomiomas em glândula adrenal, fígado, cólon e membrana epidural. O linfoma testicular também está relacionado ao VEB nesses pacientes.
A infecção pelo VIH predispõe a várias condições neoplásicas, principalmente ao linfoma não-Hodgkin e ao sarcoma de Kaposi. O potencial oncogênico do VIH está relacionado com o grave comprometimento do sistema imunológico, já que o câncer está associado a uma gama de desordens imunossupressoras. Porém, parecem existir efeitos oncogênicos diretos da infecção pelo VIH, como mutagênese insercional, super-regulação de oncogenes, estimulação antigênica crônica e desregulação de citocinas. Há, também, a hipótese de que a proteína tat (uma proteína regulatória) desse vírus promoveria crescimento em lesões de sarcoma de Kaposi.3
LINFOMA NÃO-HODGKIN
O VEB está presente em cerca de um terço dos casos de linfoma não-Hodgkin em pacientes com SIDA, os quais desenvolvem linfomas agressivos. Parece estar também implicado no desenvolvimento de alguns casos de linfoma não-Hodgkin em pacientes sem imunodeficiência evidente.12
LEUCOPLASIA PILOSA ORAL
Pouco após a infecção inicial pelo VIH e anos antes que um indivíduo desenvolva SIDA, há um aumento no número de VEB, medido através da RCP, na saliva. Este vírus está relacionado à patogênese da leucoplasia pilosa oral (LPO) associada à SIDA, e sua presença nas lesões constitui critério diagnóstico. A ocorrência da LPO pode estar ligada à carga viral alta do VEB, mas o motivo exato do aparecimento das lesões é desconhecido.
Em indivíduos VEB+, a imunodeficiência resulta na reativação da replicação do vírus na orofaringe e na expansão da população de linfócitos B infectados. Nos pacientes com SIDA, o risco de se desenvolverem linfomas associados a esse vírus correlaciona-se tanto com o grau quanto com a duração da linfopenia CD4+. Entretanto, a presença de LPO nesses pacientes ou em transplantados não se correlaciona, respectivamente, nem com a contagem de linfócitos CD4+, nem com os níveis séricos de ciclosporina A.46,47
A LPO afeta cerca de 25% dos pacientes VIH+, e constitui manifestação precoce de imunossupressão, comumente a primeira, em adultos infectados por esse vírus. Ocasionalmente, ocorre em indivíduos imunossuprimidos iatrogenicamente ou, mesmo, em indivíduos saudáveis. Portanto, embora o estado imunológico deva ser importante, a patogênese da LPO pode incluir, também, fatores determinados diretamente por vírus.
Fato importante nessa doença, com provável implicação patogenética, é a presença de co-infecção pelos tipos I e II do VEB e por múltiplas cepas do mesmo e a recombinação, inter- ou intra-cepas, de seqüências únicas ou repetidas de ADN, incluindo os genes ANVEB-2 e PLM-1 , dentro do genoma desse vírus.48,49,50,51,52,53 Segundo Resnick et al.54 é necessária a replicação ativa do VEB nas células epiteliais para que a LPO se desenvolva.
A análise de biópsias de lesões de LPO negativas para o VEB sugere que tais lesões constituiriam, antes, condição propícia à expressão desse vírus que uma conseqüência da infecção pelo mesmo.46
Existem lesões VEB- similares à LPO, algumas contendo Candida albicans , outras, por exemplo, resultando de irritação mecânica. O diagnóstico de certeza da LPO só pode ser obtido através da demonstração de genes ou de produtos gênicos do VEB nas lesões. Hibridização com AVEBs não é útil, devido à ausência de uma fase de infecção latente detectável.55
PREVENÇÃO E TRATAMENTO DA INFECÇÃO PELO VEB
Estudos sobre o papel do VEB em malignidades e o desenvolvimento de métodos de triagem apropriados para a predição de linfomas e do CNF, em populações de alto risco, são muito importantes. Contudo, a demonstração da presença do vírus em uma neoplasia raramente modifica a terapia a ser instituída e, portanto, não é indicada rotineiramente. A doença linfoproliferativa pós-transplante de medula óssea constitui uma exceção, pois tem sido tratada com sucesso com linfócitos T-citotóxicos VEB-específicos do doador.56
A imunização com hemácias de carneiro leva à produção de anticorpos heterófilos, mas estes não são persistentes nem protetores. Linhagens celulares que produzem grandes quantidades de vírus são disponíveis, existindo, pois, a possibilidade de que seja produzida uma vacina inativada. Entretanto, o risco potencial de oncogênese traz problemas para a elaboração de uma vacina eficaz contra o VEB, sendo necessárias informações adicionais sobre os riscos implicados antes que experimentos clínicos mais extensos sejam realizados em seres humanos.
Anticorpos contra uma glicoproteína de superfície do VEB, a gp340 , que se liga ao receptor celular para o vírus, demonstraram possuir atividade neutralizante contra o vírus. Recente estudo em animais revelou que a vacinação com essa glicoproteína, gerada por tecnologia de ADN recombinante, protege os mesmos contra o desenvolvimento de tumores, após serem infectados pelo VEB.57 Novas tentativas com o uso de preparações vacinais adequadas para uso em seres humanos estão sendo realizadas de forma que, futuramente, possam ser usadas, por exemplo, na prevenção do LB e do CNF em populações de alto risco.
Um estudo mostrou remissão temporária, com o uso de aciclovir, de uma desordem linfoproliferativa policlonal de linfócitos B, que se desenvolveu em um paciente, após transplante renal.58,59 Por outro lado, Sullivan et al.60 sugerem que as desordens linfoproliferativas policlonais induzidas pelo VEB em indivíduos imunocomprometidos são não-produtivas e, portanto, não-susceptíveis aos efeitos antivirais dessa droga.
Aciclovir e desciclovir são capazes de reverter, temporariamente, a LPO associada ao VEB, em pacientes com infecção pelo VIH-1.54,61 Aciclovir, na dosagem de 400mg a 800mg, cinco vezes ao dia, também tem se mostrado útil no tratamento da doença crônica ativa pelo VEB.62 Entretanto, esse agente, em geral, não tem sido benéfico em pacientes com síndromes linfoproliferativas,62 além de não ter sido eficaz na erradicação da infecção latente pelo vírus.63 A ressecção cirúrgica de massas tumorais pode ser considerada nos casos em que estas são localizadas ou pouco numerosas.62 Quando possível, a terapia para as doenças linfoproliferativas deve ser dirigida no sentido de reduzir a medicação imunossupressora. Determinados métodos imuno-virológicos podem auxiliar na identificação precoce de pacientes com alto risco de desenvolver esse tipo de doença, possibilitando intervenção terapêutica oportuna.39
Em um estudo envolvendo três crianças com diagnóstico confirmado de CNF, evidenciou-se resposta clínica satisfatória, nos três casos, com um esquema quimioterápico inicial (epirrubicina-cisplatina-bleomicina), seguido de radioterapia.30
Novas terapias, incluindo o uso de IFN-a, a terapia gênica (com genes supressores tumorais) e a infusão de anticorpos monoclonais e de linfócitos T-citotóxicos VEB-específicos, estão sendo estudadas. Até o momento, entretanto, embora muitos agentes tenham revelado atividade antiviral in vitro, as indicações para o seu uso clínico, no tratamento das doenças associadas ao VEB, são limitadas.56,62
Na infecção crônica pelo VEB, têm sido propostas tentativas de fortalecimento do sistema imune, como, por exemplo, pelo uso de imunoglobulinas, já que o aumento nos níveis de certas classes de anticorpos poderia ser benéfico.58 Postula-se que, no CNF, os níveis de anticorpos neutralizantes correlacionam-se com o prognóstico.64 Porém, ensaios clínicos controlados são necessários para melhor elucidar essas questões.
REFERÊNCIAS BIBLIOGRÁFICAS
1- Pisani P, Parkin DM, Munoz N, Ferlay J. Cancer and infection: estimates of the attributable fraction in 1990 (Review). Cancer Epidemiol Biomarkers Prev 1997;6(6):387-400.
2- Alonio LV, Picconi MA, Distefano AL, Teyssie AR. Mecanismos de oncogénesis viral. Infect & Microbiol Clín 1997;9(1):7-18.
3- Kuper H, Adami HO, Trichopoulos D. Infections as a major preventable cause of human cancer. J Int Med 2000;248:171-83.
4- Epstein MA, Achong BG. Various forms of Epstein-Barr virus infection in man: established facts and a general concept. Lancet 1973;2:836-9.
5- Hollingsworth R, Lee WH. Tumor suppressor genes: new prospects for cancer research. J Nat Cancer Inst 1991;83(2):91-6.
6- Moran B, Zerler B. Interactions between cell growth-regulating domains in the products of the adenovirus E1A oncogene. Mol Cell Biol 1988;8(4):1756-64.
7- Zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst 2000;92(9):690-8.
8- Pattengale PK, Smith RW, Perlin E. Atypical lymphocytes in acute infectious mononucleosis: identification by multiple T and B lymphocyte markers. N Engl J Med 1974;291(22):1145-8.
9- Schooley RT. Epstein-Barr Virus (Infectious Mononucleosis). In: Mandell GL, Bennett, JE, Dolin R, eds. Mandell Douglas and Bennett's Principles and Practice of Infectious Diseases. 4ed. New York: Churchill-Livingstone; 1995.1364-77. p.
10- Fahraeus R, Rymo R, Rhim JS, Klein G. Morphological transformation of human keratinocytes expressing the LMP gene of Epstein-Barr virus. Nature 1990;345:447-9.
11- Henderson S, Rowe M, Gregory C et al. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B-cells from programmed cell death. Cell 1991;65:1107-15.
12- Kanavaros P, Jiwa M, Van der Valk P, Walboomers J, Horstman A, Meijer CJLM. Expression of Epstein-Barr virus latent gene products and related cellular activation and adhesion molecules in Hodgkin's disease and non-Hodgkin's lymphoma arising in patients without overt pre-existing immunodeficiency. Hum Pathol 1993;24(7):725-9.
13- Niedobitek G, Young LS. Epstein-Barr virus persistence and virus-associated tumors. Lancet 1994;343:333-5.
14- Burkitt D. A sarcoma involving the jaws in African children. Brit J Surg 1958;46:218-23.
15- Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet 1964;1:702-3.
16- Epstein MA, Henle G, Achong BG, Barr YM. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt's lymphona. J Exp Med 1965;121:761-70.
17- Henle W, Henle G. Seroepidemiology of the virus. In: Epstein MA, Achong BG, eds. The Epstein-Barr virus. New York: Springer-Verlag; 1979: 61- 78 apud Pannuti CS. Soro-epidemiologia do vírus de Epstein-Barr (VEB). Rev Saúde públ 1981;15:93-100.
18- De-Thé G, Day NE, Geser A et al. Sero-epidemiology of the Epstein-Barr virus: preliminary analysis of an international study - a review. IARC Sci Publ 1975;(11 Pt 2):3-16.
19- Nkrumah F, Henle W, Henle G, Herberman R, Perkins V, Depue R. Burkitt's lymphoma: its clinical course in relation to immunologic reactivities to Epstein-Barr virus and tumor-related antigens. J Natl Cancer Inst 1976;57(5):1051-6.
20- Henle W, Henle G, Gunvén P, Klein G, Clifford P, Singh S. Patterns of antibodies to Epstein-Barr virus-induced early antigens in Burkitt's lymphoma. Comparison of dying patients with long-term survivors. J Natl Cancer Inst 1973;50:1163-73.
21- Epstein MA, Achong BG. The relationship of the virus to Burkitt's lymphoma. In: Epstein MA, Achong BG, eds. The Epstein-Barr virus. New York: Springer-Verlag; 1979:321-37 apud Pannuti CS. Soro-epidemiologia do vírus de Epstein-Barr (VEB). Rev Saúde públ 1981; 15. 93-100. p.
22- Lai PK, Alpers MP, MacKay-Scollay EM. Development of cell-mediated immunity to Epstein-Barr herpesvirus in infectious mononucleosis as shown by leukocyte migration inhibition. Infect & Immun 1977;17(1):28-35.
23- Okano M, Thiele GM, Davis JR, Grierson HL, Purtilo DT. Epstein-Barr virus and human diseases: recent advances in diagnosis. Clin Microbiol Rev 1988;1(3):300-12.
24- Old LJ, Boyse EA, Oettgen HF et al. Precipitating antibody in human serum to an antigen present in cultured Burkitt's lymphoma cells. Proc Nat Acad Sci USA 1966;56:1699-704.
25- Old LJ, Boyse EA, Geering G, Oettgen HF. Serologic approaches to the study of cancer in animals and in man. Cancer Res 1968;28:1288-99.
26- Evans AS, Niederman JC, McCollum RW. Seroepidemiologic studies of infectious mononucleosis with EB virus. N Engl J Med 1968;279(21):1121-7.
27- Evans AS, Niederman JC. Epstein-Barr virus. In: Evans AS, ed. Viral infections of humans. New York: Plenum Press; 1976: 209-31 apud Pannuti C.S. Soro-epidemiologia do vírus de Epstein-Barr (VEB). Rev Saúde públ 1981;15:93-100.
28- De Schryver A, Friberg Jr S, Klein G et al. Epstein-Barr virus-associated antibody patterns in carcinoma of the post-nasal space. Clin Exp Immunol 1969;5:443-59.
29- Linde A. Diagnosis of Epstein-Barr virus-related diseases. Scand J Infect Dis Suppl 1996;100:83-8.
30- Paes RAP, Teixeira RAP, Barrezuela LFM, Bruniera P. Três casos de carcinoma de nasofaringe associados ao vírus Epstein-Barr (EBV) em crianças: aspectos clínicos, patológicos e imunohistoquímicos. J Bras Patol 1996;32(2):76-82.
31- Raab-Traub N, Flynn K. The structure of the termini of the Epstein-Baar virus as a marker of clonal cellular proliferation. Cell 1986;47:883-9.
32- Evans AS, Gutensohn NM. A population-based case-control study of EBV and other viral antibodies among persons with Hodgkin's disease and their siblings. Int J Cancer 1984;34:149-57.
33- Weiss LM, Movahed LA, Warnke RA, Sklar J. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin's disease. N Engl J Med 1989;320:502-6.
34- Pallesen G, Hamilton-Dutoit SJ, Rowe M, Young LS. Expression of Epstein-Barr virus latent gene products in tumor cells of Hodgkin's disease. Lancet 1991;337:320-2.
35- Herbst H, Steinbrecher E, Niedobitek G et al. Distribution and phenotype of Epstein-Barr virus-harboring cells in Hodgkin's disease. Blood 1992;80(2):484-91.
36- Khan G, Norton AJ, Slavin G. Epstein-Barr virus in Hodgkin's disease. Relation to age and subtype. Cancer 1993;71:3124-9.
37- Abreu ES, Ferreira FVA, Rocha Filho FD et al. Doença de Hodgkin infanto-juvenil no Estado do Ceará e sua relação com o vírus de Epstein-Barr: parâmetros clínicos e análises morfológica, imuno-histoquímica e por hibridização in situ. J Bras Patol 1997;33(4):178-84.
38- Henle W, Henle G. Epstein-Barr virus-specific serology in immunologically compromised individuals. Cancer Res 1981;41:4222-5.
39-Rooney CM, Loftin SK, Holladay MS, Brenner MK, Krance, RA, Heslop HE. Early identification of Epstein-Barr virus-associated post-transplantation lymphoproliferative disease. Br J Haematol 1995;89:98-103.
40- Santos OLR, Rodrigues AG, França ER, Coelho APG, Carneiro SCS. Os herpesvírus humanos no curso da síndrome da imunodeficiência adquirida. An Bras Dermatol 1998;73(supl 2):10-8.
41- Golden MP, Kim S, Hammer SM et al. Activation of human immuno-deficiency virus by herpes simplex virus. J Infect Dis 1992;166:494-9.
42- Cohen JI. Epstein-Barr virus lymphoproliferative disease associated with acquired immunodeficiency. Medicine 1991;70:137-60.
43- Ballerini P, Gaidano G, Gong J et al. Molecular pathogenesis of HIV-associated lymphomas. AIDS Res Hum Retroviruses 1992;8:731-5.
44- Bacchi CE, Bazan R, Padovani EG, Rabenhorst SH, Bacchi MM. Linfoma do sistema nervoso central: associação com o vírus de Epstein-Barr. J Bras Patol 1996;32(3):103-9.
45- Hochberg FH, Miller G, Schooley RT, Hirsch MS, Feorino P, Henle W. Central nervous system lymphoma related to Epstein-Barr virus. N Engl J Med 1983; 309(13):745-8.
46- Reichart PA, Langford A, Gelderblom HR, Pohle HD, Becker, J, Wolf, H. Oral hairy leukoplakia: observations in 95 cases and review of the literature. J Oral Pathol Med 1989;18:410-5.
47- Schmidt-Westhausen A, Gelderblom HR, Neuhaus P, Reichart PA. Epstein-Barr virus in lingual epithelium of liver transplant patients. J Oral Pathol Med 1993; 22:274-6.
48- Patton DF, Shirley P, Raab-Traub N, Resnick L, Sixbey JW. Defective viral DNA in Epstein-Barr virus-associated oral hairy leukoplakia. J Virol 1990; 64(1):397-400.
49- Walling DM, Edmiston SN, Sixbey JW, Abdel-Hamid M, Resnick L, Raab-Traub N. Coinfection with multiple strains of the Epstein-Barr virus in human immunodeficiency virus-associated hairy leukoplakia. Proc Natl Acad Sci USA 1992;89:6560-4.
50- Walling DM, Raab-Traub N. Epstein-Barr virus intrastrain recombination in oral hairy leukoplakia. J Virol 1994;68(12):7909-17.
51- Walling DM, Perkins AG, Webster-Cyriaque J, Resnick L, Raab-Traub N. The Epstein-Barr virus EBNA-2 gene in oral hairy leukoplakia: strain variation, genetic recombination, and transcriptional expression. J Virol 1994;68:7918-26.
52- Miller WE, Edwards RH, Walling DM, Raab-Traub N. Sequence variation in the Epstein-Barr virus latent membrane protein 1. J Gen Virol 1994;75:2729-40.
53- Walling DM, Clark NM, Markovitz DM et al. Epstein-Barr virus coinfection and recombination in non-human immunodeficiency virus-associated oral hairy leukoplakia. J Infect Dis 1995;171:1122-30.
54- Resnick L, Herbst JS, Ablashi DV et al. Regression of oral hairy leukoplakia after orally administered acyclovir therapy. JAMA 1988;259(3):384-8.
55- Niedobitek G, Young LS, Lau R et al. Epstein-Barr virus infection in oral hairy leukoplakia: virus replication in the absence of a detectable latent fase. J Gen Virol 1991;72:3035-46.
56- Papadopoulos EB, Ladanyi M, Emanuel D et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogenic bone marrow transplantation. N Engl J Med 1994;330:1185-91.
57- Morgan AJ, Allison AC, Finerty S, Scullion FT, Byars NE, Epstein MA. Validation of a first-generation Epstein-Barr virus vaccine preparation suitable for human use. J Med Virol 1989;29:74-8.
58- Jones JF, Shurin S, Abramowsky C et al. T cell lymphomas containing Epstein-Barr viral DNA in patients with chronic Epstein-Barr virus infections. N Engl J Med 1988;318(12):733-41.
59- Sullivan JL, Byron KS, Brewster FE, Sakamoto K, Shaw JE, Pagano JS. Treatment of life-threatening Epstein-Barr virus infections with acyclovir. Am J Med 1982;73:262-6.
60- Sullivan JL, Baker SM, Byron KS, Brewster FE, Mulder C. Failure of acyclovir to inhibit polyclonal Epstein-Barr virus induced lymphoproliferation in the immunocompromised host. Fed Proc 1983;42:458.
61- Greenspan D, De Souza YG, Conant MA et al. Efficacy of desciclovir in the treatment of Epstein-Barr virus infection in oral hairy leukoplakia. J AIDS 1990; 3(6):571-8.
62- Cohen JI. Epstein-Barr virus infections, including infectious mononucleosis. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds. Harrison's Principles of Internal Medicine. 15th ed. (International Edition). New York: McGraw-Hill; 2001:1109-11.
63- Carvalho LHFR. Mononucleose infecciosa. In: Farhat CK, Carvalho, ES, Carvalho LHFR, Succi RCM, eds. Infectologia Pediátrica, 2ed., Rio de Janeiro: Atheneu; 1999:447-59.
64- Pearson GR, Neel HB, Weiland LH et al. Antibody-dependent cellular cytotoxicity and disease course in North American patients with nasopharyngeal carcinoma: a prospective study. Int J Cancer 1984;33:777-82.
Fonte:http://rmmg.org/artigo/detalhes/1534#:~:text=O%20v%C3%ADrus%20Epstein%2DBarr%20(VEB,e%20outros%20tipos%20de%20neoplasia.
Revisão de Literatura Associação do EBV com Tumores
Sólidos
Artigo submetido em 28/2/05;
aceito para publicação em 9/8/05
Associação do vírus Epstein-Barr (EBV) com
tumores sólidos
Association of Epstein-Barr virus (EBV) with Solid
Tumors
Marcos Antonio Pereira de Lima 1, Silvia Helena
Barem Rabenhorst2
Resumo
O gama-herpes
vírus Epstein-Barr (EBV) é um
vírus ubíquo, que estabelece infecção persistente em mais
de 90% da população mundial adulta. O EBV
está associado a várias desordens proliferativas benignas e malignas
de origem linfóide, tais como mononucleose
infecciosa, linfoma de
Burkitt, doença de Hodgkin
e doença linfoproliferativa pós- transplante, nas quais o seu papel
oncogênico tem sido
largamente estudado. Em tumores sólidos, a relação do EBV é
bem documentada
em carcinomas de nasofaringe. Contudo, novos achados
têm demonstrado que o EBV apresenta
um espectro de infecção celular mais amplo do que se conhecia, sendo detectado em células de
outros tipos de tumores sólidos,
com evidências que apontam para
a sua participação na tumorigênese dessas neoplasias.
O presente artigo inicia com uma breve
descrição da biologia
do EBV e tem como objetivo
realizar uma compilação dos achados encontrados na literatura, concernentes à associação do EBV a neoplasias sólidas, apontando as evidências do envolvimento viral e as características das infecções que
são encontradas em cada tecido.
Palavras-chave: : Vírus de
Epstein-Barr; Tumores sólidos; Oncogênese.
Abstract
The gamma-herpes Epstein-Barr virus (EBV) is a ubiquitous viral agent that causes persistent infection in more than 90% of the world adult population. EBV is
associated with several lymphoproliferative disorders such as infectious mononucleosis, Burkitt's lymphoma, Hodgkin's disease, and post-transplant lymphoproliferative disease. The oncogenic role
of EBV
in tumors of lymphoid origin has
been widely investigated. With
respect to solid tumors, the role of EBV
is well documented in nasopharyngeal carcinoma. However, new findings have shown that the
spectrum of cellular infection of EBV is
broader, having been detected in cells from other kinds of
solid tumors, thus indicating its involvement
in tumorigenesis of these tumors. The current article begins
with a brief description of EBV biology and aims to provide a summary of new findings concerning the association between EBV and solid
tumors, highlighting evidence of
the viral role and characteristics
of infections in each tissue.
Key words: Epstein-Barr virus; Solid tumors; Oncogenesis.
Departamento de Microbiologia Médica - Universidade
Federal do Ceará.
1 Mestrando do Curso de
Pós-Graduação em Microbiologia Médica - Universidade Federal do Ceará.
2 Professora de Genética Molecular do Departamento de Patologia e Medicina Legal -
Faculdade de Medicina -
Universidade Federaldo Ceará.
Endereço
para correspondência:Silvia Helena Barem Rabenhorst - Rua Marcos Macedo,
1301, Apt,: 802 - Aldeota - Fortaleza -
CE CEP.: 60150-190 tel.: (085) 3267-3840 /
(085) 9994-5689. E-mail: silviarabenhorst@yahoo.com.br
INTRODUÇÃO
O vírus
Epstein-Barr (EBV) é um gama-herpes vírus constituído
de DNA linear fita dupla, envolvido por um capsídeo icosadeltaédrico composto de 162 capsômeros,
recoberto por um envelope glicoprotéico.1 É
amplamente distribuído no mundo e estima-se que
mais
de 90% da população
mundial adulta está infectada
por esse vírus.2
O EBV é transmitido pela saliva, infectando inicialmente as células epiteliais
da orofaringe, nasofaringe e
glândulas salivares, por receptores
ainda não identificados, onde
freqüentemente ocorre replicação.
Posteriormente, os vírus alcançam
tecidos linfóides adjacentes e infectam linfócitos
B através da ligação
entre a glicoproteína viral gp350/220
e o receptor CD 21 (CR2) do componente C3d
do sistema complemento.3 Após
essa associação, o vírus
penetra nos linfócitos B por fusão do envoltório com a membrana celular
e o capsídeo é então liberado no citoplasma. O genoma antes linear é transportado
para o núcleo tornando-se circular, permanecendo em estado latente, sob a
forma de DNA epissomal extracromossômico.4
O genoma viral apresenta
um comprimento de cerca de
172 Kpb, o qual foi dividido em
regiões, com base
na posição no mapa de restrição
da endonuclease BamHI (Bacillus amyloliquefaciens H), e nomeadas
de acordo com o
tamanho do fragmento, indo de A
à Z, sendo
o fragmento BamHI A o maior deles.5
Devido a estímulos ainda
não totalmente esclarecidos,
o EBV pode iniciar um ciclo lítico. O primeiro gene a ser expresso nesse ciclo
é o BZLF1 (BamHI Z left-ward
open reading frame 1), também conhecido
como ZEBRA.6 Esse gene codifica
uma proteína que apresenta
função de ativador transcricional, iniciando
uma cascata de eventos que determinam a expressão de algumas proteínas, tais como os antígenos precoces (EAs), BHRF1 (BamHI H right-ward open reading frame 1) e BCRF1 (BamHI C right-ward open reading frame 1), incluindo a expressão da DNA polimerase
viral .7
Durante as infecções latentes, é possível verificar
a expressão de alguns genes virais que codificam: seis proteínas
conhecidas como antígenos nucleares do EBV, EBNA-1, 2, 3A, 3B (também conhecido EBNA-4), 3C
e LP (também designada EBNA-5); três proteínas latentes de membrana LMP-1,
LMP-2A e LMP-2B;
duas pequenas moléculas de RNA não-poliadeniladas,
EBER- 1 e 2; alguns transcritos
com múltiplos splices, da região BamHI A do genoma viral, BARF0
(BamHI
A right- ward open reading
frame) e BARF1.7,8,9
Esses genes, no entanto, não são
expressos concomitantemente nas diversas células de tecidos ou linhagens
tumorais EBV-positivas, sendo proposta a existência de quatro tipos de
latência, com padrões distintos de expressão dos genes virais, os quais estão
descritos na tabela 1.
De acordo
com a International Agency for Research
on Cancer - IARC, o Epstein-Barr
Vírus é classificado como um carcinógeno do grupo I.
Não obstante, diversos autores
investigam a função das
proteínas virais expressas
e a participação dessas no desenvolvimento das neoplasias. Alguns estudos
tentam correlacioná-las com a superexpressão
ou mesmo inibição de proteínas celulares envolvidas
em processos oncogênicos. A
seção a seguir descreve alguns achados relevantes relativos aos genes
latentes do EBV.
GENES LATENTES: MECANISMOS TUMORIGÊNICOS
A oncoproteína EBNA-1, codificada pelo gene homônimo, é responsável pela manutenção do genoma viral,
já que ao se ligar à região
oriP permite a replicação pela DNA polimerase celular.10 Atua, também, como
fator de transcrição do gene viral LMP-15 e supressor, em nível transcricional,
da expressão do oncogenec-erbB-2 celular.11 O EBNA-2, por
sua vez,
transativa genes virais,
tais como LMP-1, LMP-2 e genes
celulares como CD21, CD23 e
o c-myc.12,13 Também, induz a proliferação/ transformação de linfócitos B.14,15 Ademais, interage com o fator transcricional RBP-Jk, regulando
positivamente sua atividade.16 Juntamente com
EBNA-LP, são as primeiras
proteínas a serem observadas, sendo essas suficientes para induzir
a transição da fase G0 para
G1 do ciclo celular em
linfócitos-B, possivelmente por
aumentarem
a expressão de Ciclina D2.5,17
A família
EBNA-3 parece ter
importante papel na transformação de células B.10 Podem induzir a expressão de genes virais (LMP-1) e celulares (CD21) e se
combinar com a
proteína Rb, favorecendo a transformação
celular.5 Os EBNA-3 também se associam à
proteína RBP-Jk
, mas ao contrário da EBNA2 regulam negativamente sua atividade.16
Em relação
aos EBERs, tem sido relatado que esses conferem
resistência à apoptose em células de linfoma de Burkitt
e tumorigenicidade em camundongos SCID. Induzem, ainda, a transcrição de IL-10 em células de Linfoma de Burkitt.18 Niller et al19 demonstraram
uma região de ligação para a proteína c-Myc, cerca de 130pb fita acima
do gene EBER-1, o qual,
por sua vez, tem
sido demonstrado ser capaz de
bloquear os efeitos
pró- apoptóticos da proteína c-Myc.15
 |
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Tabela 1. Genes expressos do EBV quanto aos tipos
de latência e tecido associado.
Tabela 1. Genes expressos do EBV quanto aos tipos
de latência e tecido associado.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Referências 3,7,8,10,18,26,27,29
Referências 3,7,8,10,18,26,27,29
Alguns efeitos da LMP-1,
ainda, são motivo de
controvérsias entre alguns autores. De acordo com Dolcetti et al20,
a LMP-1 demonstra um efeito
imortalizador sob os linfócitos, decorrente da indução da expressão do oncogene bcl-2, correlação
essa não observada por outros
autores.21,22 Entretanto, a LMP-1 atua
como membro da família de receptores TNF, ativando o fator transcricional NF-kB, induzindo a expressão de receptores
celulares (CD40, CD23, EGFR), moléculas de adesão (CD57,
ICAM-1, LFA 1 e 3) e citocinas
(IL-6 e IL-10).10,23,24. Já
a LMP-2A regula negativamente a atividade de proteínas tirosino-quinases citoplasmáticas (PTK), tais como as da família Syk. Promove a
sobrevivência de linfócitos B e inibe a reativação
viral.5,10,25 Também, pode induzir
transformação em células
epiteliais.5
O BARF1 é descrito como
gene latente
exclusivamente em neoplasias de origem epitelial, sendo
descrito como um gene lítico precoce nas
infecções de linfócitos-B. Atua como homólogo funcional do receptor do fator estimulador de colônia 1
(CSF-1) humano.10 Exerce efeitos imortalizantes
em células epiteliais primárias humanas in vitro e
induz tumorigenicidade em linhagens de fibroblastos.9
METODOLOGIA
Os artigos utilizados nesta revisão foram selecionados
nos portais CAPES, PUBMED e SCIELO e obtidos na integra nos referidos portais ou solicitados por meio da BIREME, quando não disponíveis
na Internet. Todos foram organizados
de acordo com os sítios
tumorais e sintetizados para compilação das informações,
as quais foram então discutidas na atual revisão.
TÉCNICAS
DE DETECÇÃO
As técnicas utilizadas para
demonstrar o EBV em espécimes tumorais
e que,
subseqüentemente, permitiram
analisar a participação do referido vírus no desenvolvimento
das várias neoplasias associadas, incluem: amplificação de seqüência do genoma viral
através de reação em cadeia da polimerase (PCR); hibridização
in
situ ( HIS), utilizando
sondas complementares tanto na seqüência do genoma viral, quanto
nos segmentos de RNAm viral; imuno- histoquímica e imunofluoscência, utilizando anticorpos monoclonais específicos para proteínas virais expressas; Western blotting;
e Southern blot.3,8,26
A técnica de Southern blot
é, também, aplicada para
avaliar a
clonalidade do EBV encontrado nas neoplasias, a
presença de infecção lítica e investigar a
ocorrência de integração do genoma viral ao genoma da célula hospedeira.8,27 A clonalidade do vírus é verificada através da
manutenção do número de repetições de 500pb, descritas
como TR (tandem repeat),
que se encontram nas extremidades
do genoma viral. Dentro
deste conceito, a verificação de uma única banda de mais de
8,0 Kpb indica a presença de epissomos com
mesmo número de TR, sugerindo um padrão monoclonal
do genoma viral.3 Contrariamente, a constatação de bandas de mais
de 8,0 Kpb dispostas em escada aponta para um padrão policlonal. Adicionalmente, a demonstração
de bandas de cerca de 4,0 Kpb indica a presença de genoma linear e subseqüentemente, infecção lítica.8
A associação do EBV a neoplasias de origem linfóide
tem sido
amplamente estudada. Contudo, os relatos da presença
do referido vírus em neoplasias sólidas
têm sido cada vez mais freqüentes. Segue abaixo, uma revisão da literatura existente até o
momento, nos portais PUBMED, CAPES e SCIELO, concernente à associação do EBV em tumores sólidos.
EBV EM TUMORES SÓLIDOS
Carcinoma
de Nasofaringe
O Carcinoma de Nasofaringe (CNF) foi a primeira neoplasia
de origem epitelial a ser associada ao EBV, relatada por Wolf et al28,
em 1973, e, desde
então, tem sido vastamente
estudada no mundo. Até o momento dessa revisão, foi possível
encontrar mais de
700 artigos, associando CNF e EBV no portal
PUBMED. Nesses tumores, o percentual de associação com o EBV parece não apresentar variações em todas partes do mundo.8 Virtualmente, todos os casos
de CNF indiferenciados (também descritos como linfoepitelial ou linfoepitelioma) são EBV-positivos, sendo o referido
vírus pouco observado em tipos diferenciados.23
Níveis plasmáticos
elevados de DNA viral são
observados em pacientes de estágio inicial
e avançado de CNF, sendo sugerido que a quantificação
do DNA do EBV pode ser útil no diagnóstico e monitoramento como,
também, um indicador
de risco para recorrência.29
Nesse tipo tumoral, o EBV desenvolve uma
latência do tipo II (tabela 1) e o genoma viral obtido apresenta caráter monoclonal.23
A presença do vírus, em
todas as células tumorais
(detectado pela técnica de hibridização
in situ), aliado ao elevado
percentual de associação e a
monoclonalidade, é uma das principais indicações do papel
tumorigênico do EBV em CNF.30 Sarac et al21
demonstraram uma correlação positiva entre a expressão de
LMP-1 e bcl-2, sendo que a expressão
de bcl-2 não foi dependente da expressão de
LMP-1. Adicionalmente, Niemhoma
et al.31, verificaram uma superexpressão de bcl-2 e
p53 em casos EBV-positivos, sugerindo que o vírus possa estar envolvido
na regulação desses dois oncogenes, atuando
como um importante fator etiológico
nos CNF.
Carcinoma Gástrico
Além da função bem estabelecida do Helicobacter pylori, a participação do EBV no desenvolvimento de
carcinoma gástrico (CG) tem
sido recentemente demonstrada. A associação
do vírus Epstein-Barr aos CGs apresenta uma distribuição mundial,
variando de 2 - 18%.32 No Japão, o percentual
de associação é de 7%, na Rússia
8,7%, na Alemanha 18%, no México 8,2% e nos Estados Unidos 16%.33
Segundo Hausen et al9, o EBV também está associado a cerca de
80-100% dos carcinomas gástricos do
tipo linfoepitelial.
Os carcinomas gástricos EBV-positivos desenvolvem uma latência do tipo I (tabela 1). Apesar da presença da LMP-2A não ser característica desse tipo de latência, pouca quantidade pode ser expressa em determinadas situações.3,9
É interessante notar que o gene LMP-1, um
dos principais oncogenes conhecidos
do EBV, não é expresso
nesse tipo de carcinoma.
No entanto, recentemente foi
demonstrado que outro gene viral, o BARF1,
expresso nesse tipo de neoplasia,
exerce efeitos imortalizantes em células
epiteliais in vitro, o que
pode indicar uma via alternativa
na oncogênese de carcinomas gástricos mediada pelo
EBV.9
A indicação de que o EBV esteja relacionado ao processo oncogênico
nos CGs, assim como
nos CNFs, é baseada no significativo percentual de associação com esses tumores, bem como na presença do
vírus em quase todas
as células tumorais dos carcinomas gástricos EBV- positivos e o caráter monoclonal dessas células. Contudo, Hausen et al34 não
constataram EBV em lesões pré- neoplásicas, sugerindo a ocorrência da infecção viral após o desenvolvimento
da neoplasia.
Alguns estudos apontam
para ocorrência de metilação
de supressores tumorais
induzida pelo EBV como um dos mecanismos virais nesse processo3.5,36,37
Um achado interessante vem dos
estudos de Chong et al37, no qual demonstraram
que apesar da elevada freqüência de metilação
encontrada nos genes p14, p15, p16, TIMP-
3, E-caderina, DAPK, GSTPi e MGMT em CGs EBV- positivos
observaram a supressão da expressão de DNA metiltransferases, uma enzima responsável pela metilação
do DNA, sugerindo uma via alternativa do vírus para metilação desses genes.
Neoplasias
da Musculatura Lisa
A presença do EBV,
também, foi relatada
em leiomiomas e leiomiossarcomas, em pacientes imunossuprimidos.3 Inicialmente, o EBV foi demonstrado nesses tumores,
em crianças aidéticas, através da técnica de HIS por MacClain et al38 Contudo, a análise da clonalidade do genoma viral por Southern blot
revelou um caráter policlonal,
sendo também observados elevados níveis
de receptores CD21 na
superfície das células tumorais. Adicionalmente,
Lee et al27, estudando três casos de tumor de músculo-liso após transplante, verificaram a expressão de
EBNA-2, EBER-1 e ausência de LMP-1 e de receptores CD21. Nesse
estudo, a demonstração da expressão de uma das principais oncoproteínas
virais, o EBNA-2, e a origem monoclonal do genoma viral
são evidências da participação do EBV no desenvolvimento desse tipo
de neoplasia.
Apesar dos estudos contraditórios quanto à presença do receptor CD21, a participação
do EBV parece estar associada à imunossupressão, já que pacientes
imunocompetentes não desenvolvem leiomiossarcoma EBV-positivo.3
Carcinoma
Intracervical
Apesar
da infecção por Papilomavírus Humanos ( HPV),
especialmente pelos tipos
16 e 18, ser considerada atualmente o principal fator de risco para o
desenvolvimento de Carcinoma Intracervical, alguns estudos
têm demonstrado que o EBV possa,
também, ter participação nesse processo. Landers et al39,
realizando HIS-DNA e PCR, verificaram a presença de EBV em 43% dos carcinomas cervicais estudados
e uma menor associação em lesões pré-malignas NIC-II
e NIC- III, não sendo detectado
em lesões NIC-I e tecidos normais. Recentemente, Shimakage et al12 também observaram a presença do EBV em carcinoma cervical, neoplasia
cervical intra-epitelial (NIC-I, II,
III) e ausência em tecido
normal, utilizando as técnicas de imunofluorescência indireta para o EBNA-2 e de
hibridização in situ com
sondas complementares ao RNAm de EBNA-2 e
de BamHI-W. Esse último foi, também,
detectado em casos de cervicite crônica,
sugerindo que a expressão de BamHI W isoladamente não seja
capaz de induzir o desenvolvimento de neoplasias cervicais. Em contrapartida, Hording et al40,
fazendo uso da técnica de
PCR, investigaram a presença de genoma dos vírus HPV (16, 18) e EBV em espécimes de pacientes da Groelândia e
Dinamarca, obtendo positividade apenas para HPV.
Mesmo com os poucos estudos, Shimakage et al12 defendem que o EBV é um fator de risco para o desenvolvimento
de carcinoma cervical, independente
do HPV. Ademais, a demonstração da
expressão de EBNA-2 é um importante achado relativo
ao papel do EBV na tumorigênese cervical, considerando alguns dos
efeitos descritos dessa proteína.
Carcinoma
Hepatocelular
Apesar de algumas controvérsias, alguns
estudos demonstram que além do
vírus da hepatite B (HBV), o vírus Epstein-Barr pode, também, estar associado à gênesis do Carcinoma Hepatocelular (CHC).
Sugawara et al26 detectaram a presença do genoma viral em 13/ 35 (37%) dos espécimes
clínicos provenientes de pacientes japoneses
submetidos à hepatectomia, com expressão de
BARF0 e EBNA1 em
apenas um pequeno percentual de células tumorais
(7- 13%) e ausência de EBNA-2, -3, -LP, LMP-1, LMP-2A
e LMP-2B.
Os EBERs que são
normalmente expressos nos
quatro padrões de latência (tabela 1),
também não foram detectados nas amostras positivas para o EBV. Foi verificado,
ainda, o caráter monoclonal do
genoma viral, bem como um elevado percentual de associação
do EBV com o vírus da hepatite C (9 dos 13 casos EBV- positivos),
sugerindo um sinergismo entre
esses vírus no desenvolvimento do carcinoma hepatocelular.
Recentemente, Li et al41 verificaram a presença da LMP-1
em células tumorais de 15/78 casos
de pacientes
chineses, através de imuno-histoquímica. Nos dois referidos estudos,
não foi verificada correlação significativa entre o EBV e
o HBV.
Contrariamente, em estudos realizados na
Holanda, Hausen et al42 detectaram fraca positividade para o
fragmento BamHI W do EBV, pela
técnica de PCR- ensaio imunoenzimático em 5 das 16 amostras
estudadas. Não foram observadas
as expressões de EBNA-1, BARF1, LMP-1, EBERs e ZEBRA, o que,
segundo os autores, se contrapõem ao papel do EBV na oncogênese
hepática, levando a acreditar que o resultado
fracamente positivo para
o DNA viral, fosse decorrente
de amplificação a partir de genoma viral
presente em linfócitos infiltrados. Junying et
al43, analisando espécimes de CHC oriundos
da Alemanha e Reino Unido, e Akhter et al44
da América do Norte, não detectaram a presença de EBV. Nesse
último estudo,
apenas HBV e/ou HCV (vírus da hepatite
C) foram detectados durante a investigação.
No entanto, Chu et al45,
analisando 41 espécimes obtidos de
pacientes norte-americanos, demonstraram
quatro casos positivos. Dois foram evidenciados
pela presença de EBER1 e ZEBRA, mas restritos a raros linfócitos do infiltrado. Os outros dois casos
envolviam as células
tumorais e foram caracterizados
pela presença de EBNA-1 e ausência
de EBER-1, EBNA-4, LMP-1 e ZEBRA, ocorrendo
em pacientes de etnia asiática. Esses achados
corroboram com Sugawara et al26, e apontam para
a possibilidade de indivíduos asiáticos serem mais suscetíveis
a desenvolver CHC, quando infectados pelo EBV, o qual
atuaria como co-fator nesse processo.
Câncer da Mama
Os
primeiros a relatarem a
presença do EBV, em câncer da mama,
foram Labrecque et al46, os quais utilizando a técnica de PCR,
amplificando as regiões
BamHI W e BamHI C, obtiveram 19/91 (21%) casos positivos.
Subseqüentemente, empregando as técnicas de HIS, nos 19 casos positivos,
obtiveram 12 (63%)
casos com células tumorais marcadas, quando
utilizando sondas complementares à
seqüência de DNA viral e 6
(31,5%) casos, quando utilizando sondas para EBER-1.
Bonnet et al47, através de PCR, demonstraram a presença de EBV em 51 de 100 casos de carcinoma
invasivo primário da mama. Tomando alguns espécimes
positivos, puderam constatar através de imuno- histoquímica,
a expressão de EBNA-1 em uma fração de células
tumorais e a ausência da expressão de EBERs,
através da técnica de HIS. Recentemente, Grinstein et al48
observaram a presença de EBV em 14/33 (42%)
carcinomas, através de PCR e imuno-histoquímica, caracterizados também
pela expressão de EBNA-1 e ausência de LMP-1,
ZEBRA e EBER-1.
Adicionalmente, o EBV foi detectado em lesões pré-malignas, o que
aponta para a participação do vírus na carcinogênese mamária. Tanto Bonnet et al47 como
Grinstein et al48 não analisaram
a clonalidade do genoma viral, o que
poderia fornecer mais evidências referentes ao papel do EBV no desenvolvimento
desses tumores. Mais
recentemente, Ribeiro-Silva et al49, empregando imuno-
histoquímica,
demonstraram positividade para EBNA-
1 em 32/85 (37,6%) casos de carcinoma da
mama em pacientes brasileiras, sendo mais freqüentemente associado a carcinomas
ductais pouco diferenciados. Não sendo observada a expressão de EBNA-1 em
fibroadenomas e tecidos normais.
Os estudos de Chu et al50 e Herrmann
& Niedobitek51
são contrários aos anteriormente citados. Esses autores, fazendo
uso das técnicas de HIS-EBER e imuno-
histoquímica para EBNA-1,
não detectaram os referidos expressos
virais em nenhum dos espécimes
estudados, sugerindo que os dois
referidos genes não estivessem sendo expressos.
Adicionalmente, Herrmann & Niedobitek51 obtiveram
positividade em quatro dos 59 casos estudados, fazendo uso da técnica
de PCR. Contudo, nesse caso é possível
que a positividade tenha sido decorrente
de amplificação, a partir de linfócitos infectados presentes no infiltrado, e ao considerar o elevado percentual
de positividade observado por Bonnet et al47, é possível que esse último evento também tenha
ocorrido em seus experimentos.
Câncer de Pulmão
O EBV
tem sido também associado a carcinoma pulmonar do tipo linfoepitelioma (LELC). As primeiras evidências
de sua participação decorrem de estudos
de Pittaluga et al52, em cinco pacientes chineses, nos quais
foi demonstrada a presença do EBV, através da técnica de
HIS, apenas em células
epiteliais. Sendo essa infecção de caráter monoclonal.
Corroborando
esses achados, Wong et al53 observaram a presença de EBV (através de HIS-EBER) de caráter monoclonal em nove dos
167 casos (5,4%) de carcinoma
pulmonar do tipo linfoepitelioma em pacientes
chineses. Ademais, foram
observadas a presença de LMP-1 e ausência de EBNA-2 em 4 dos
9 casos, através da técnica
de imuno-histoquímica. Recentemente,
Grinstein et al48, estudando
54 casos de carcinomas, observaram positividade para EBNA-1 em quatro
casos (7,4%), sendo a positividade restrita
a apenas uma fração das células tumorais (5-30%).
Contrariamente, Castro et al54,
utilizando a técnica de hibridização in situ para EBERs,
não demonstraram EBV em nenhum dos seis casos de LELC obtidos de pacientes ocidentais, sugerindo que
o EBV é mais freqüentemente demonstrado em casos de câncer pulmonar de indivíduos asiáticos. Entretanto, o "n" estudado pelo referido autor
foi
insuficiente, considerando que em estudos prévios
o EBV demonstrou baixa freqüência
de associação.
Carcinoma de Glândula Salivar
A presença
do EBV tem sido também demonstrada em carcinoma de
glândula salivar. Raab-Traub et al30 verificaram, através da técnica
de HIS, a expressão de RNAm de LMP-1, BARF-0 e EBER-1 em carcinoma indiferenciado de glândulas parótidas, limitado
ao tecido
tumoral. É importante ressaltar, que o EBV é freqüentemente observado
nesse sítio desenvolvendo infecção lítica, contudo, não foi verificada a presença de formas lineares. Posteriormente, Leung et al55
relataram HIS-EBER fortemente positiva e presença de LMP-1,
através de imuno-histoquímica, em
10/10 (100%) casos de carcinoma
linfoepitelial de glândulas salivares. Em ambos os estudos, o caráter monoclonal
do genoma viral foi demonstrado.
Carcinoma
Oral
O EBV, também, tem sido demonstrado
em carcinoma oral de células escamosas.56-61 Na maioria
desses, foi verificado
um elevado percentual de associação, variando de 25 a 86%, utilizando a técnica de PCR.
Entretanto, Higa et al14 verificaram
a expressão de LMP-1, EBNA-2 (através de
imuno-histoquímica) e EBER1 (através de HIS) em 49/95 (51%) casos de pacientes japoneses. Enquanto, Kobayashi et al62 e Gonzalez-Moles et al63 demonstraram a presença
da LMP- 1, através de
imuno-histoquímica, respectivamente em
6/46 (13%) e 15/78 (19,2%) dos casos
estudados. Em ambos trabalhos,
a HIS-EBER foi negativa.
Contudo, Shimakage et al64
verificaram a expressão de EBER1 em 16/24 (66,6%) casos,
através de HIS.
Achados conflitantes vêm do estudo de Cruz et al61, que, analisando 36 casos PCR-positivo,
constataram a não expressão dos
transcritos EBNA-1, EBNA-2, LMP-
1, LMP-2, BHRF1 e BARF0, através da técnica de RT- PCR. As reações
de imuno-histoquímica para ZEBRA,
EBNA-1 e LMP-1, também foram
negativas.
Apesar de
controversos, os estudos denotam que
o EBV pode estabelecer infecção latente com expressão de genes latentes em células de carcinoma oral, indicando que o mesmo possa participar
no desenvolvimento deste tipo neoplásico ou, como sugerido por Sand et al59, atuar junto de fatores de risco conhecidos, tais como fumo e consumo de álcool.
Outras
Neoplasias Associadas
Leung et al65 verificaram a
expressão de EBER, através de HIS, em
sete de 29 casos de carcinoma sinonasal
de pacientes chineses, sendo que dois dos sete casos
positivos também expressavam LMP-1. Esses mesmos autores descreveram a presença do vírus em um caso de carcinoma indiferenciado de glândula
lacrimal.66 Shimakage et
al67 relataram a expressão de EBNA-2,
LMP-1 e EBERs em 27/27 (100%) de tumores
testiculares. Grinstein et al48 demonstraram o EBV em um dos 19 casos de carcinoma
de cólon, com uma fração
de células tumorais expressando EBNA-1.
Também, verificaram EBV em sete de 19 amostras de câncer prostático de diversos
graus de malignidade, com núcleos
das células tumorais fortemente marcados. Kekis et al68 relataram a
presença de EBV, através da técnica de HIS- EBER, em
apenas um caso de
carcinoma linfoepitelial pancreático.
No estudo realizado por Leung et al69, 2/7 casos
de carcinoma de ouvido médio
(respectivamente, carcinoma
de células escamosas não-queratinizado e carcinoma
indiferenciado) foram positivos para
EBER. A análise, através de imuno-histoquímica, demonstrou ausência de LMP-1 e EBNA-2,
ambos os casos apresentavam
histologia compatível com o tipo
linfoepitelioma.
CONCLUSÕES
A associação do vírus
Epstein-Barr a muitas das neoplasias sólidas, tem sido demonstrada
em estudos recentes, o que justifica
a pouca quantidade de
literatura e as evidências algumas vezes inconclusivas a respeito da participação viral no processo oncogênico
de alguns tumores. Entretanto, a
verificação da clonalidade do EBV, na maioria
desses tumores, sugere que as infecções tenham precedido o processo tumorigênico, apontando fortemente para a participação do vírus nesse processo.
A detecção de EBV policlonal
ou a expressão parcial de antígenos virais, em apenas uma fração das células, podem sugerir mecanismos distintos no processo tumorigênico.
Bonnet et al47 e Hausen et al34
atribuem a expressão de EBNA-1,
em apenas uma fração
de células tumorais, relatada em alguns trabalhos, a uma baixa expressão dessa proteína ou uma menor sensibilidade
da técnica por alguns autores. Entretanto,
como discorrido por Ambinder70, esse fenômeno pode ser decorrente da perda do
genoma viral durante o processo tumorigênico,
decorrente de um mecanismo viral do
tipo "hit and run".
Um achado interessante
refere-se a maior freqüência
de alguns tipos tumorais EBV-positivos em indivíduos de etnia asiática, sugerindo que fatores comportamentais e genéticos contribuam para o
desenvolvimento dessas neoplasias.
A imunossupressão, assim como nas doenças linfoproliferativas pós-transplante (DLPT) e linfomas não-Hodgkin, em pacientes aidéticos, parece favorecer
o desenvolvimento de neoplasias sólidas associadas ao EBV, particularmente os leiomiomas e leiomiossarcomas, permitindo a disseminação do vírus para diversos tecidos, bem como a expressão de
vários genes latentes
nas células portadoras.
A diversidade de padrões de expressão de genes latentes observados denota a complexidade da infecção pelo
EBV. Evidências da heterogeneidade
de expressão do EBV associadas ao tecido infectado vêm com os
estudos de Sugawara et al26 e Chu et al45
referentes a CHC e os
de Bonnet et al47 e Grinstein et al48, relativos a câncer da mama.
Os achados desses autores apresentaram
características comuns: a presença de EBNA-1 e ausência de EBERs e apontam para a
possibilidade de que o EBV estabeleça
uma infecção latente com padrão
semelhante nesses dois tipos
neoplásicos, diferindo de todos os
padrões já conhecidos, pela ausência de
EBERs.
Devido a essa diversidade de expressão, é possível que o vírus
esteja associado a neoplasias de
outros sítios, mas não
tenha sido detectado pela falta de expressão do alvo da técnica empregada. Em
alguns estudos, observa- se a necessidade de mais de uma abordagem técnica para confirmação da freqüência do EBV. A presença do vírus nos infiltrados linfocíticos, também, deve
ser considerada quando se usa a
técnica de PCR. Essa técnica,
entretanto, torna-se uma ferramenta importante para detecção
em tumores, em que não se
tem conhecimento do padrão
de latência. Associada à hibridização
in situ e à imuno-histoquímica, pode
posteriormente confirmar e
estabelecer o padrão de latência. O comportamento do EBV e o mecanismo
pelo qual contribui para o seu desenvolvimento
em algumas neoplasias, ainda requerem
mais estudos, mas certamente possuem participação relevante.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Kieff E. Epstein-Barr virus and its replication. In: Fields BN, Knipe DM, Howley
PM. Fundamental Virology.
3th ed. Philadelphia: Lippincott-Raven; 1996. p. 1109-63.
2. Callan MF. The immune
response to Epstein-Barr virus. Microbes Infect. 2004;6(10):937-45.
3. Tsuchiya S. Diagnosis of Epstein-Barr virus-associated
diseases. Crit Rev Oncol Hematol. 2002;44(3):227-38.
4. Hsieh WS, Lemas MV, Ambinder RF. The
biology of Epstein-Barr virus in post-transplant lymphoproliferative disease.
Transpl Infect Dis. 1999;1(3):204-12.
5. Murray PG, Young LS. Epstein-Barr virus infection: basis
of malignancy and potential for therapy. Expert Rev Mol Med. 2001;15:1-20.
6. Gulley ML. Molecular diagnosis of Epstein-Barr virus- related diseases. J Mol Diagn.
2001;3(1):1-10.
7. Okano M. Haematological associations of Epstein-Barr virus infection. Baillieres
Best Pract Res Clin Haematol. 2000;13(2):199-214.
8. Ambinder RF, Mann RB. Detection and characterization of Epstein-Barr virus in clinical
specimens. Am J Pathol. 1994;145(2):239-52.
9. zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer
CJ, van den Brule AJ.
Unique transcription pattern of
Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer
Res. 2000;60(10):2745-8.
10. Ohga S, Nomura A, Takada H, Hara T. Immunological aspects of
Epstein-Barr virus infection. Crit Rev Oncol Hematol. 2002;44(3):203-15.
11. Chuang TC, Way TD, Lin YS, LeeYC, Law SL, Kao MC. The Epstein-Barr
virus nuclear antigen-1 may act as a
transforming suppressor of the HER2/neu oncogene. FEBS Lett. 2002;532:135-42
12. Shimakage M, Sasagawa T. Detection of Epstein--Barr virus-
determined nuclear antigen-2
mRNA by in situ hybridization. J Virol Methods.
2001;93(1-2):23-32.
13. Kaiser C, Laux G, Eick D, Jochner
N, Bornkamm GW, Kempkes B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear
antigen 2. J Virol. 1999;73(5):4481-4.
14. Higa M, Kinjo T, Kamiyama K, K, Iwamasa T, Hamada T, Iyama K. Epstein-Barr virus (EBV) subtype in EBV related oral squamous cell carcinoma in Okinawa,
a subtropical island, in southern
Japan, compared with Kitakyushu and Kumamoto
in mainland Japan. J Clin Pathol.
2002;55:414-23.
15. Niller HH, Salamon D, Ilg K, Koroknai
A, Banati
F, Schwarzmann F, et al. EBV-associated neoplasms: alternative
pathogenetic pathways. Med
Hypotheses. 2004;62(3):387-91.
16. Cludts I, Farrell PJ. Multiple functions within the Epstein- Barr virus EBNA-3A protein. J Virol. 1998;72(3):1862-9.
17. Knecht
H, Berger
C, al-Homsi AS, McQuain C, Brousset
P. Epstein-Barr virus oncogenesis. Crit Rev Oncol Hematol. 1997;26(2):117-35.
18. Takada K, Nanbo A. The role of EBERs in oncogenesis. Semin Cancer Biol. 2001;11(6):461-7.
19. Niller HH, Salamon D, Ilg K, Koroknai
A, Banati F, Bauml
G, et al. The in vivo binding
site for oncoprotein c-Myc in the promoter for Epstein-Barr virus (EBV) encoding RNA (EBER) 1 suggests
a specific role for
EBV in lymphomagenesis. Med Sci Monit.
2003;9(1):HY1-9.
20. Dolcetti R, Boiocchi M. Epstein-Barr vírus in the pathogenesis of Hodgkin's disease. Biomed Pharmacother.
1998;52:13-25.
21. Sarac S, Akyol U, Kanbur B, Pouraz A, Akyol G, Yilamz T, et al.
Bcl-2 and LMP1 Expression in Nasopharyngeal Carcinomas. Am J Otolaryngol. 2001; 22(6):377-82.
22. Tao Q, Srivastava G, Loke SL, Ho FC. Lack of correlation between expression of Epstein-Barr
virus (EBV) latent membrane
protein and bcl-2 oncoprotein in vivo. J Clin Pathol. 1994;47(7):589-91.
23. Raab-Traub N. Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol. 2002;12(6):431-41.
24. Niedobitek G. Epstein-Barr
virus infection in the pathogenesis
of nasopharyngeal carcinoma. Mol Pathol. 2000;53(5):248-54.
25. Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system.
Nat Rev Immunol. 2001 Oct;1(1):75-82.
26. SugawaraY, MizugakiY, Uchida T, Torii T, Imai S, Makuuchi M, et
al. Detection of Epstein-Barr virus (EBV) in hepatocellular
carcinoma tissue: a novel EBV latency characterized by the absence of EBV-encoded small RNA expression.
Virology. 1999 Apr 10;256(2):196-202.
27. Lee ES, Locker J, Nalesnik M, Reyes J, Jaffe R, Alashari M, et
al. The association of Epstein-Barr virus with smooth-
muscle tumors occurring after organ transplantation. N Engl J
Med. 1995 Jan 5;332(1):19-25.
28. Wolf H, zur
Hausen H, Becker V. EB viral genomes
in epithelial nasopharyngeal carcinoma cells. Nature. 1973;244:245-7.
29. Fan H, Gulley ML. Epstein-Barr viral load measurement as a
marker of EBV-related disease. Mol
Diagn. 2001 Dec;6(4):279-89.
30. Raab-Traub N, Rajadurai P, Flynn K, Lanier AP. Epstein- Barr virus infection in carcinoma
of the salivary
gland. J Virol. 1991 Dec;65(12):7032-6.
31. Niemhom S, Kitazawa S, Murao S, Kunachak S, Maeda S. Co-expression of p53 and bcl-2 maycorrelate to
the presence of
epstein-barr virus genome and the expression
of proliferating cell nuclear antigen in nasopharyngeal carcinoma. Cancer
Lett. 2000 Nov 28;160(2):199-208.
32. Chang MS, Kim HS, Kim CW, Kim YI, Lan Lee B, Kim WH. Epstein-Barr virus, p53 protein, and microsatellite instability in the adenoma-carcinoma
sequence of the stomach.
Hum Pathol. 2002 Apr;33(4):415-20.
33. Koriyama C, Akiba S, Iriya K, Yamaguti T, Hamada GS, Itoh T, et al. Epstein-Barr Virus-associated Gastric Carcinoma
in Japanese Brazilians
and Non-Japanese Brazilians. Jpn J Cancer Res. 2001 Sep;92:
911-7.
34. zur
Hausen A, van Rees BP,
van Beek J, Craanen ME, Bloemena E, Offerhaus GJ, et al. Epstein-Barr virus in gastric carcinomas and gastric stump carcinomas: a late event in
gastric carcinogenesis. J Clin Pathol. 2004 May;57(5):487-91.
35. Kang GH,LeeS,Kim WH,Lee HW,Kim JC, Rhyu MG,et al. Epstein-Barr virus-Positive Gastric Carcinoma Demonstrates Frequent Aberrant Methylation
of Multiple Genes and Constitutes CpG Island Methylator Phenotype-Positive Gastric Carcinama. Am J
Pathol. 2002;160(3):787-94.
36. Osawa T, Chong
JM, Sudo M, Sakuma K, Uozaki
H, Shibahara J, et al. Reduced expression and promoter methylation of p16 gene in Epstein-Barr virus-associated
gastric carcinoma. Jpn J Cancer
Res. 2002 Nov;93(11):1195-200.
37. Chong JM, Sakuma K,
Sudo M, Ushiku T, Uozaki H,
Shibahara J, et al. Global and
non-random CpG-island methylation in gastric
carcinoma associated with Epstein-
Barr virus. Cancer Sci. 2003 Jan;94(1):76-80.
38. McClain KL, Leach CT, Jenson HB, Joshi VV, Pollock BH,
Parmley RT, et al. Association of Epstein-Barr virus with leiomyosarcomas in children
with AIDS. N Engl J Med.
1995 Jan 5;332(1):12-8.
39. Lander RJ, O'Leary JJ, Crowley M, Healy I, Annis
P, Burke L, et al. Epstein-Barr virus in normal,
pre-malignant, and malignant lesions of the uterine
cervix. J Clin Pathol.
1993 Oct;46(10):931-5.
40. Hording U, Daugaard S, Bock JE. Human papillomavirus,
Epstein-Barr virus, and cervical
carcinoma in Greenland. Int J Gynecol
Cancer. 1992 Nov;2(6):314-7.
41. Li W, Wu BA, ZengYM, Chen GC, Li XX, Chen JT, et al. Epstein-Barr virus in hepatocellular carcinogenesis. World J
Gastroenterol. 2004;10(23):3409-3413.
42. zur Hausen A, van Beek J, Bloemena E, ten Kate FJ, Meijer CJ, van den BruleAJ. No role for Epstein-Barr virus in Dutch hepatocellular carcinoma: a study
at the DNA, RNA and
protein levels. J Gen Virol.
2003 Jul;84(Pt 7):1863-9.
43. Junying J, Herrmann
K, Davies
G, Lissauer D, Bell A, Timms
J, et al. Absence
of Epstein-Barr virus DNA in the tumor cells of European hepatocellular carcinoma. Virology.
2003 Feb 15;306(2):236-43.
44. Akhter S, Liu H, Prabhu R, DeLucca C, Bastian
F, Garry RF, et al.
Epstein-Barr virus and human hepatocellular
carcinoma. Cancer Lett. 2003 Mar 20;192(1):49-57.
45. Chu PG, Chen YY, Chen W, Weiss LM. No direct role for Epstein-Barr virus in American
hepatocellular carcinoma.
Am J Pathol. 2001 Oct;159(4):1287-92.
46. Labrecque LG, Barnes DM, Fentiman IS,
Griffin BE. Epstein-Barr virus in epithelial cell tumors: a breast cancer study.
Cancer Res. 1995
Jan 1;55(1):39-45.
47. Bonnet M, Guinebretiere JM, Kremmer
E, Grunewald V, Benhamou
E, Contesso G, et al. Detection
of Epstein-Barr virus
in invasive breast cancers.
J Natl Cancer Inst.
1999 Aug 18;91(16):1376-81.
48. Grinstein S, Preciado MV, Gattuso P, Chabay PA, Warren WH, De Matteo
E, et al. Demonstration of Epstein-Barr
virus in carcinomas
of various sites. Cancer Res. 2002 Sep 1;62(17):4876-8.
49. Silva AR, Ramalho LN, Garcia SB, Zucoloto
S. Does the correlation between EBNA-1 and p63 expression in breast carcinomas provide a clue to tumorigenesis in Epstein-Barr virus-related breast malignancies?Braz J Med Biol Res. 2004 Jan;37(1):89-95.
50. Chu JS, Chen CC, Chang KJ. In situ detection of Epstein-
Barr virus in breast cancer. Cancer Lett.
1998 Feb 13;124(1):53-7.
51. Herrmann
K, Niedobitek G. Lack of
evidence for an association
of Epstein-Barr virus infection
with breast carcinoma. Breast Cancer
Res. 2003;5(1):13-7.
52. Pittaluga S, Wong MP, Chung LP, Loke SL. Clonal Epstein-
Barr virus in lymphoepithelioma-like carcinoma of the lung. Am J Surg Pathol.
1993; 17(7):678-82.
53. Wong MP, Chung LP, Yuen ST, Leung SY, Chan SY, Wang
E, et al. In situ detection
of Epstein-Barr virus in non-small cell lung carcinomas. J Pathol.
1995;177(3):233-40.
54. Castro CY, Ostrowski ML, Barrios R,
Green LK, Popper
HH, Powell S, et al. Relationship between Epstein-Barr virus
and lymphoepithelioma-like carcinoma
of the lung: a clinicopathologic study of 6 cases and review of the literature.
Hum Pathol. 2001;32(8):863-72.
55. Leung SY, Chung LP, Yuen ST, Ho CM, Wong MP, Chan
SY.
Lymphoepithelial carcinoma of the salivary
gland: in situ detection of Epstein-Barr virus. J Clin Pathol. 1995 Nov;48(11):1022-7.
56. Mao E, Smith
C. Detection of Epstein-Barr virus (EBV)
DNA by the polymerase chain reaction (PCR) in oral smears
from healthy individuals and patients
with squamous cell carcinoma. J Oral Pathol Med. 1993;22:12-7.
57. D'Costa J, Saranath D, Sanghvi
V, Mehta A. Epstein-Barr virus in tobacco-induced oral cancers and oral lesions
in patients from India.
J Oral Pathol Med. 1998;27:78-82.
58. Gonzalez-Moles MA, Galindo
P, Gutierrez
J, Rodriguez- Archilla A, Ruiz-Avilla I, Sanchez-Fernandez E. Expression
of the p53 protein in oral squamous cell carcinomas associated with
Epstein-Barr virus. Microbios.
2000;102(403):147-54.
59. Sand
LP, Jalouli J, Larsson PA,
Hirsch JM. Prevalence
of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen
planus, and normal oral mucosa.
Oral Surg Oral Med Oral Pathol Oral
Radiol Endod. 2002 May;93(5):586-92.
60. Szkaradkiewisz A, Kruk-Zagajewska A, Wal M, Jopek A, Wierzbicka M, Kuch A. Epstein-Barr virus and human papillomavirus infections and oropharyngeal squamous cell
carcinomas. Clin Exp Med. 2002 Nov;2(3):137-41.
61. Cruz I, Van Den BruleAJ, BrinkAA, Snijders PJ, Walboomers JM, Van Der Waal I, et al.
No direct role for Epstein-Barr virus in oral carcinogenesis: a study at the DNA, RNAand
protein levels. Int J Cancer. 2000;86:356-61.
62. Kobayashi I, Shima K, Saito I, Kiyoshima T, Matsuo
K, Ozeki S, et al. Prevalence of Epstein-Barr virus in oral
squamous cell carcinoma. J Pathol. 1999;189:34-9.
63. Gonzalez-Moles MA, Gutierrez J, Rodriguez MJ, Ruiz- Avila I, Rodriguez-Archilla A Epstein-Barr
virus latent membrane protein-1 (LMP-1) expression in oral squamous cell carcinoma. Laryngoscope. 2002 Mar;112(3):482-7.
64. Shimakage M, Horii K, Tempaku A, Kakudo K, Shirasaka
T, Sasagawa T. Association of Epstein-Barr virus with oral cancers.
Hum Pathol. 2002 Jun;33(6):608-14.
65. Leung SY, Yuen ST, Chung
LP, Kwong
WK, Wong MP, Chan SY. Epstein-Barr virus is present in a wide histological
spectrum of sinonasal carcinomas. Am J Surg Pathol.
1995 Sep;19(9):994-1001.
66. Leung SY, Chung LP, Ho CM, Yuen ST, Wong MP, Kwong
WK. An Epstein-Barr virus positive undifferentiated carcinoma in the
lacrimal sac. Histopathology. 1996 Jan;28(1):71-5.
67. Shimakage M, Oka T, Shinka T, Kurata A,
Sasagawa T, Yutsudo M. Involvement of Epstein-Barr virus expression
in testicular tumors.
J Urol. 1996 Jul;156(1):253-7.
68. Kekis PB, Murtin C, Kunzli BM, Kappler
A, Buchholz B, Buchler MW,
et al. Epstein-Barr virus-associated lymphoepithelial carcinoma
in the pancreas. Pancreas. 2004 Jan;28(1):98-102.
69. LeungSY,Yuen ST, Ho CM, KwongWK, ChungLP. Presence of Epstein-Barr virus in lymphoepithelioma-like carcinoma of the middle ear. J Clin Pathol. 1998;51:602-605.
70. Ambinder RF. Gammaherpesviruses and "Hit-and-Run" oncogenesis. Am J Pathol.
2000 Jan;156(1):1-3.
Fonte:https://rbc.inca.gov.br/index.php/revista/article/view/1912/1163
J Câncer 2020; 11(7):1737-1750. doi:10.7150/jca.37282 Esse assunto
Análise
Linfomas e cânceres epiteliais associados ao vírus Epstein Barr em humanos
Richmond Ayee 1,2 , Maame Ekua Oforiwaa Ofori 1 , Edward Wright 3 , Osbourne Quaye 1,2![]()
1. Departamento de Bioquímica, Biologia Celular e Molecular, Universidade de Gana, Legon, Accra, Gana
2. Centro da África Ocidental para Biologia Celular de Patógenos Infecciosos (WACCBIP), Universidade de Gana, Legon, Accra, Gana
3. Departamento de Bioquímica, Universidade de Sussex, Brighton, Reino Unido
Ayee R, Ofori MEO, Wright E, Quaye O. Epstein Barr Virus Associado Linfomas e cânceres de epitélio em humanos. J Câncer 2020; 11(7):1737-1750. doi:10.7150/jca.37282. Disponível em https://www.jcancer.org/v11p1737.htm
Resumo
O vírus Epstein Barr (EBV) é um vírus oncogênico cosmopolita, infectando cerca de 90% da população mundial e está associado a tumores originados tanto de epitélios quanto de células hematopoiéticas. A transmissão do vírus ocorre principalmente por meio de secreções orais; no entanto, a transmissão através de transplante de órgãos e transfusão de sangue tem sido relatada. Para evitar o reconhecimento imunológico, o EBV estabelece uma infecção latente nos linfócitos B, onde expressa conjuntos limitados de proteínas chamadas de programas de transcrição de EBV (ETPs), incluindo seis antígenos nucleares (EBNAs), três proteínas latentes de membrana (LMP) e RNA não traduzido chamado EBV. RNA codificado (EBER), mostrou transformar eficientemente células B em células linfoblásticas. Esses programas sofrem diferentes padrões de expressão que determinam a ocorrência de distintos tipos de latência na patogênese de um determinado tumor. Os tumores derivados de células hematopoiéticas incluem, mas não se limitam a, linfoma de Burkitt, linfoma de Hodgkin, distúrbios linfoproliferativos pós-transplante e linfoma de células T/NK (natural killer). O EBV sofre infecção lítica em células epiteliais para amplificação da partícula viral para transmissão onde expressa genes em estágio lítico. No entanto, por razões ainda não reveladas, o EBV passa da expressão de genes do estágio lítico para a expressão de ETPs em células epiteliais. A expressão dos ETPs leva à transformação de células epiteliais em células de proliferação permanente, resultando em malignidades derivadas de células epiteliais, como câncer de nasofaringe, câncer gástrico, e câncer de mama. Nesta revisão, resumimos as atualizações atuais sobre malignidades epiteliais e derivadas de células B associadas ao EBV, e o papel dos produtos do gene de latência do EBV na patogênese dos cânceres, e sugerimos áreas para estudos futuros ao considerar medidas terapêuticas
Palavras -chave : vírus Epstein Barr, linfomas, câncer epitelial, programa de latência
Introdução
O vírus gamaherpes humano 4 (HHV-4) ou vírus Epstein Barr (EBV) é um vírus oncogênico ubíquo pertencente à família Herpesviridae [ 1 , 2 ], que é ainda classificada nas subfamílias alfa, beta e gamaherpes vírus. O EBV é um membro clássico da subfamília Gammaherpesvirinae e entre nove vírus que foram identificados por infectar apenas humanos [ 3 , 4 ]. O vírus foi descoberto e isolado em células do linfoma de Burkitt Africano por Epstein Barr e Achong em 1964 [ 5 , 6 ], e foram relatados para estabelecer infecção assintomática latente em cerca de 90% da população humana adulta do mundo [ 7 ]]. Fatores socioeconômicos e de desenvolvimento têm demonstrado influenciar a idade em que a infecção primária pode ocorrer. Por exemplo, em países da África Subsaariana onde o padrão de vida é ruim, a infecção primária ocorre na primeira infância e a maioria das crianças infectadas soroconverte aos 3 anos de idade, enquanto em países desenvolvidos ou ricos, a infecção primária é adiada até o final da infância ou jovem idade adulta [ 8 ]. Para estabelecer a infecção primária, o vírus é transmitido por via oral onde exibe duplo tropismo ao infectar dois alvos fisiológicos principais, células epiteliais e linfócitos B [ 3 ]. Além de infectar o epitélio e as células B, o vírus também demonstrou infectar alvos não naturais, como linfócitos T e células natural killer (NK) .].
A replicação lítica do vírus ocorre nas células epiteliais, mas o vírus pode estabelecer latência infectando células B encontradas nos tecidos linfoides da faringe do anel de Waldeyer [ 7 , 10 ]. Ao entrar nas células B, o genoma viral se integra ao genoma do hospedeiro e persiste como um provírus [ 11 ] ou permanece no núcleo como um epissoma circular não integrado e expressa um conjunto restrito de genes que impulsionam a latência e a sobrevivência do hospedeiro célula [ 12 , 13 ]. A expressão dos genes do estágio de latência, conhecidos como programas de latência, nas células B levam a linfomas derivados de células B como resultado da transformação das células em linhas linfoblásticas (Figura 1). O vírus pode ser reativado a partir da latência nas células B por um mecanismo que ainda não foi elucidado. Em indivíduos imunocompetentes, os títulos virais são controlados por células T citotóxicas específicas do EBV [ 14 ]. Embora o EBV sofra replicação lítica nas células epiteliais, onde os genes do estágio lítico são expressos, o vírus pode mudar para a expressão dos genes do estágio de latência e levar à transformação das células epiteliais em células de proliferação permanente e resultando em malignidades derivadas das células epiteliais. Figura 1 ) [ 15 ].
Nesta revisão, resumimos as atualizações atuais sobre malignidades epiteliais e derivadas de células B associadas ao EBV e o papel dos produtos do gene de latência do EBV na patogênese dos cânceres. Além disso, sugerimos áreas que podem ser exploradas por pesquisadores em estudos futuros ao considerar medidas terapêuticas contra essas malignidades.
Estrutura do genoma do EBV
O genoma do EBV é um DNA linear de fita dupla, aproximadamente 172 Kbp com mais de 85 genes codificadores de proteínas (Open reading frames (ORF)) [ 16 , 17 ]. As ORFs codificam proteínas envolvidas na regulação da replicação do DNA e expressão gênica e manutenção da integridade do genoma em células filhas [ 18 ]. As nomenclaturas das ORFs foram determinadas com base no mapa de fração de restrição Bam HI em que os genes estavam em ordem decrescente de acordo com seus tamanhos [ 19]. Os genes codificadores de proteínas são divididos em genes líticos e latentes que desempenham papéis estruturais e não estruturais. O genoma viral é ainda dividido em domínios de sequência únicos longos e curtos que são separados por uma série de repetições diretas terminais de 0,5 Kbp que são encontradas no final de cada domínio de sequência [ 20 ]. As repetições diretas terminais aumentam a capacidade de codificação do genoma e servem como biomarcador para determinar a origem progenitora do EBV nas células infectadas [ 17 , 21 ].
Transformação de linfócitos B e células epiteliais em células malignas pelo vírus Epstein Barr (EBV) . Os epitélios e os linfócitos B são transformados pelo EBV em células malignas como resultado da expressão dos produtos do gene de latência do EBV.
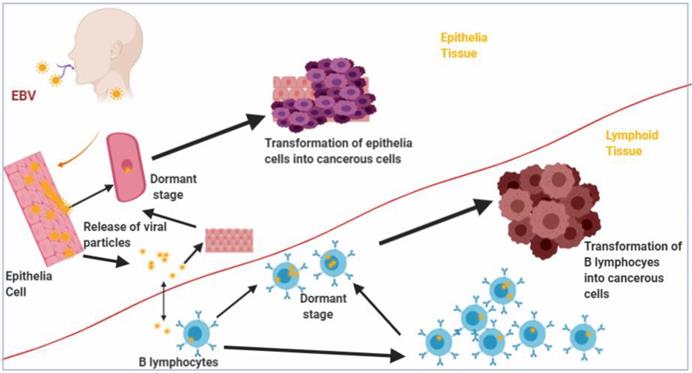
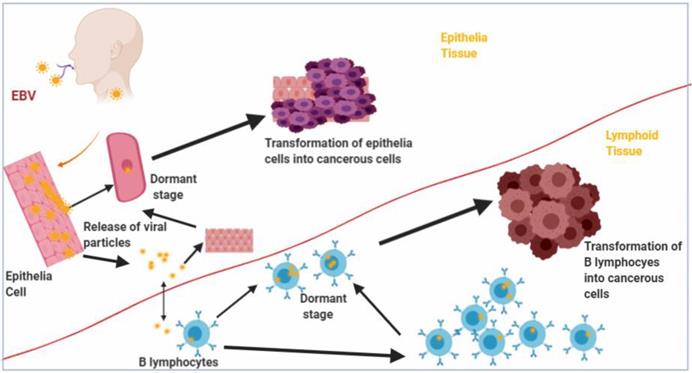{kind=link}
Classificação e distribuição geográfica do EBV
O EBV pode ser classificado em dois genótipos principais, A e B, ou tipos 1 e 2, com base na divergência de sequência nas proteínas nucleares, Epstein Barr Nuclear Antigen (EBNA)-1, -2, -3A, -3B e 3C [ 22 ]. No entanto, EBNA-2 é geralmente usado para a classificação porque a proteína tem a menor porcentagem de homologia de sequência entre os dois genótipos [ 23 , 24 ]. Os dois genótipos de EBV ocorrem em todo o mundo, mas variam em distribuição geográfica; O EBV tipo 1 é encontrado globalmente, mas predominante em populações americanas, chinesas, europeias e do Sudeste Asiático (SEA), enquanto o tipo 2 é predominantemente encontrado na África .]. Os dois genótipos também variam em propriedades biológicas; O EBV tipo 1 é mais eficiente na imortalização de células B e o tipo 2 tem maior capacidade lítica [ 26 , 27 ]. O vírus é classificado em sete cepas diferentes: China 1 (C1), China 2 (C2), China 3 (C3), Mediterrâneo com (Med+) ou sem deleções (Med-), Alaskan (AL) e Carolina do Norte, com base no polimorfismo de nucleotídeos, deleções de 30 pb ou alterações de aminoácidos de assinatura no gene LMP1 em comparação com a sequência de um protótipo de EBV, B95-8 [ 22 , 28 ]. As diferentes cepas apresentam diversidade na atividade tumorigênica; uma deleção de 30 pb no terminal carboxílico, por exemplo, tem uma maior atividade tumorigênica e baixa imunogenicidade em comparação com a variante não deletada [29 ].
Atividades biológicas de produtos de genes de latência codificados por EBV
O estabelecimento de infecção latente pelo EBV tem sido implicado em várias malignidades [ 30 ] devido à expressão de conjuntos limitados de proteínas latentes, que desempenham vários papéis biológicos discutidos na Tabela 1 .
Atividades biológicas de produtos gênicos do estágio de latência do vírus Epstein Barr e cânceres associados
| Proteína de latência EBV | Tipo de latência | Atividade biológica | Cânceres associados d |
|---|---|---|---|
| EBNA- 1a | Latência I, II, III | Segregação do genoma viral em progênies, replicação de DNA, inibição do MHC classe I, aumenta a degradação de p53 | Linfoma de Burkitt, Câncer gástrico, Câncer de mama |
| EBNA-2 | Latência III | Upregulation de proteínas do hospedeiro e virais (transativação), facilitar a imortalização de células B | Doença linfoproliferativa pós-transplante |
| EBNA-3 | Latência III | Transativação de transcrição de proteínas hospedeiras e virais, imortalização de células B | Doença linfoproliferativa pós-transplante |
| EBNA-LP b | Latência III | Transativação de EBNA-2 para inativar supressores tumorais, essencial para imortalização de células B | Doença linfoproliferativa pós-transplante |
| LMP-1/ 2c | Latência II/III | Sobrevivência de células B, regulação positiva de proteínas antiapoptóticas, imita a sinalização associada ao ligante CD 40, ativa constitutivamente o crescimento e as vias de sinalização que promovem a sobrevivência celular | Linfoma de Hodgkin, Câncer de nasofaringe, Distúrbio linfoproliferativo pós-transplante, Linfoma de células T/NK, Câncer de mama |
| EBV-Micro RNAs | Latência I, II, III | mRNAs do hospedeiro alvo envolvidos na apoptose, proliferação e transformação. Suprimir a apresentação de antígenos e ativação de células imunes | Câncer gástrico, linfoma de células T/NK, câncer de nasofaringe |
a EBNA-1 é expresso e detectado em todas as malignidades associadas ao EBV. b EBNA-LP também é conhecido como EBNA-5. c LMP-1/2 estão envolvidos em tumores de epitélio e células B, no entanto, LMP 2 é frequentemente detectado na maioria de todos os tumores em comparação com LMP-1. d Os tumores associados não se limitam apenas aos discutidos nesta revisão.
Antígenos nucleares de Epstein Barr (EBNAs)
Seis EBNAs (1, 2, 3A, 3B, 3C e LP) são expressos durante a infecção latente em células hospedeiras, e suas funções biológicas foram relatadas por pesquisadores em todo o mundo [ 31 , 32 ]. A Figura 2 mostra as atividades biológicas dos antígenos nucleares do EBV (EBNA) na tumorigênese. A expressão desses antígenos varia ao longo dos programas de latência estabelecidos pelo EBV nas células infectadas. Durante a latência III, todos os seis EBNAs são expressos, porém na latência II e I, apenas um EBNA (EBNA-1) é expresso, mas nenhuma proteína EBNA é expressa durante a latência 0 [ 8 , 32 ]. Todos os EBNAs estão envolvidos na regulação da transcrição e também participam da ativação transcricional dos genes LMP virais ( LMP-1 e -2) em células infectadas [ 33 , 34 ].
EBNA-1 é uma proteína de ligação de DNA específica de sequência que interage com três sítios alvo de sequência palindrômica única (família de elementos repetidos (FR), elemento de simetria de díade (DS) e sequências encontradas a jusante do promotor Q (Qp)), que são repetidas várias vezes no genoma viral [ 35 ]. O EBNA-1 liga-se aos elementos FR, que atuam como potenciadores do promotor viral C, para direcionar a transcrição de todos os seis EBNAs. Enquanto o envolvimento dos elementos DS pelo EBNA-1 leva à regulação da replicação do DNA viral associada à fase S, a interação com Qp regula negativamente a transcrição do EBNA-1 dirigido por Qp [ 36 , 37 ]. O EBNA-1 também garante a segregação do genoma viral em células filhas e regula positivamente o promotor LMP, sustentando a sobrevivência celular ou a imortalização de células infectadas.32 ]. Um estudo mais recente relatou que o EBNA-1 derivado do câncer de nasofaringe é necessário para a manutenção do epissoma do EBV e da replicação do DNA durante a infecção latente [ 38 ]. A proteína EBNA-1 é composta por terminais N e C que são separados um do outro por repetições variáveis de sequências de glicina-alanina. As repetições da sequência de aminoácidos ocultam o EBNA-1 do reconhecimento imunológico por um inibidor de ação cis do complexo principal de histocompatibilidade (MHC) classe I e impede a apresentação de antígenos pela via ubiquitina-proteossomo [ 39 , 40 ].
Ao contrário do EBNA-1, que se liga ao DNA, o EBNA-2 não interage com o DNA, mas envolve fatores de transcrição celular, como CBF1/RBP-Jk, PU.1 e outras proteínas para regular a expressão de genes virais e celulares. 41 ]. Os genes celulares que são regulados positivamente pelo EBNA-2 incluem CD 23 e c-myc encontrados em células B. A ativação de c-myc leva à expressão de proteínas como ciclinas do tipo D e ciclina E, que estão associadas à divisão celular [ 42 ]. O EBNA-2 também interage com outros fatores de transcrição, como fatores de ligação Cp, LMP-1 e -2 para regulação positiva de genes virais que são responsáveis pela imortalização de células B [ 41 , 43 , 44 ].
Actividades biológicas dos antigénios nucleares do EBV (EBNAs) na tumorigénese . EBNA-1 inibe a apresentação de antígeno pelo complexo principal de histocompatibilidade (MHC) classe I; EBNA-1 e EBNA-2 ativam a expressão de LMP-1; EBNA-2 e EBNA-3 interagem com C-myc que ativa constitutivamente a ciclina D/E levando à proliferação celular desregulada; A EBNA-LP pode ativar diretamente a ciclina D/E e a proteína quinase dependente de DNA (DNA-PKCs) para promover a proliferação celular; A EBNA-LP promove a sobrevivência celular interagindo com a proteína 1 específica da célula hematopoiética antiapoptótica (HS-1) associada à proteína X-1 (HAX-1).
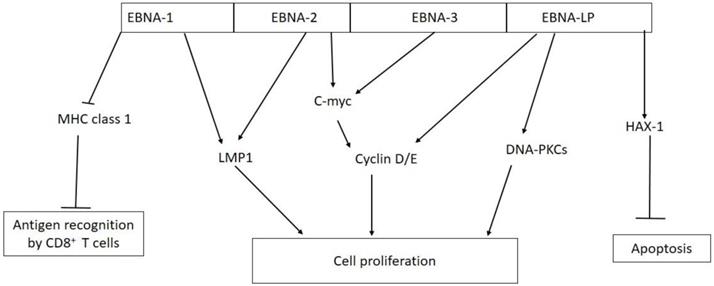
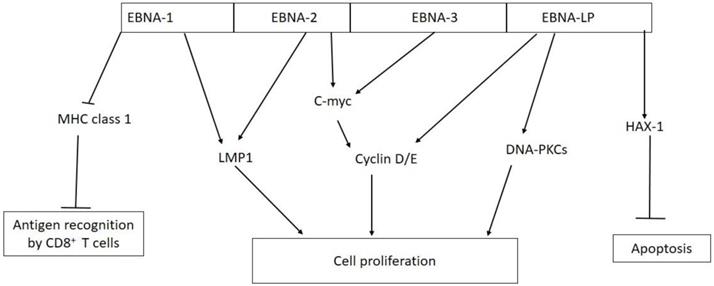{kind=link}
A família EBNA-3 de proteínas nucleares consiste em três grandes fosfoproteínas nucleares, EBNA-3A, -3B e -3C [ 44 ]. Todas as proteínas EBNA-3 compartilham poucas homologias de sequência na região terminal N, com o domínio conservado usado para envolver fatores de transcrição celular, como CBF1/RBP-Jk [ 45 ]. Ao se ligar ao CBF1/RBP-Jk, o EBNA-3 atua como um repressor da transativação mediada pelo EBNA-2 de proteínas celulares e virais [ 46 ]. Estudos genéticos in vitro mostraram que apenas EBNA-3A e -3C são necessários para imortalização de células infectadas por EBV [ 47 , 48]. Embora o EBNA-3 B seja dispensável, um relatório mostrou que a proteína regula o homing das células B alterando a expressão do receptor de quimiocina 4 (CXCR4) [ 49 ].
A proteína líder do antígeno nuclear de Epstein Barr (EBNA-LP), também conhecida como EBNA-5, é uma fosfoproteína nuclear que é co-expressa com EBNA-2 após infecção por EBV de células-alvo [ 50 ]. A co-expressão de EBNA-LP e EBNA-2 demonstrou aumentar a ativação transcricional de EBNA-2 de proteínas celulares e virais [ 44 , 51 ]. O EBNA-LP desempenha um papel fundamental na transformação de células B em um linfoblasto de proliferação permanente com base na observação de que as células que expressam os genes mutantes foram menos transformadas em comparação com as células com o gene EBNA-LP do tipo selvagem [ 52 , 53]. A proteína líder interage com proteínas celulares, algumas das quais são oncogênicas e supressoras de tumor (pRb, p53, p14ARF e Fte1/S3a), reguladoras do ciclo celular (DNA-PKcs e HA95) e uma proteína antiapoptótica (HAX-1 ) [ 54 - 56 ]. O EBNA-LP regula positivamente a expressão do gene timo e da quimiocina regulada por ativação (TARC), que foi proposto para desempenhar um papel fundamental na transformação e sobrevivência de células B [ 57 , 58 ]. Estudos anteriores relataram que EBNA-LP estimula a transativação de EBNA-2 de proteínas virais, como proteínas latentes de membrana 1 e -2B (LMP-1 e -2B) em células imortalizadas [ 51 , 59 , 60 ].
Proteína de membrana latente (LMP)
Três proteínas de membrana latentes, LMP-1-2A e -2B, são expressas pelo EBV durante a latência II e III em células infectadas com EBV [ 61 ]. Todas as três proteínas são expressas como proteínas da membrana da superfície celular e são necessárias para a sobrevivência e transformação das células infectadas em células em proliferação permanente (Figura 3 ).
O gene LMP-1 é um oncogene essencial, que é expresso como um receptor constitutivamente ativo na maioria das células tumorais associadas ao EBV [ 62 ]. LMP-1 é uma proteína de 356 aminoácidos com um domínio N-terminal citoplasmático curto, seis domínios transmembrana e um domínio citoplasmático C-terminal longo de 200 aminoácidos [ 63 ]. O domínio N-terminal amarra a LMP-1 na superfície da membrana plasmática, e a oligomerização, bem como a auto-agregação da proteína, é mediada pelos seis domínios transmembrana. O C-terminal é a região biologicamente ativa de LMP-1, com dois domínios funcionais, a saber, C-terminal Activating Regions (CTAR)-1 e -2, que são necessários para uma eficiente transformação de células B mediada por EBV [ 64]. Relatórios anteriores mostraram que esses dois domínios funcionais interagem com proteínas de sinalização intracelular, como fator associado ao receptor do fator de necrose tumoral (TNF) (TRAF)-1, -2, -3, -4, -5 e -6 encontrados no tumor via de sinalização do receptor do fator de necrose (TNFR) em células B e células epiteliais [ 65 , 66 ]. Ao interagir com essas moléculas adaptadoras, o CTAR ativa constitutivamente as principais vias de sinalização, como a quinase regulada por sinal extracelular (ERK), proteínas quinases ativadas por mitógeno (MAPKs), PI3K/Akt, AP-1, Jun N-terminal protein kinase (JNK ), as vias p38 e NF-κB canônica e não canônica e JAK/STAT [ 67 - 69]. A ativação dessas principais vias de sinalização resulta na regulação positiva da expressão de proteínas antiapoptóticas, como A 20, Bcl-2, e regulação negativa da proteína supressora de tumor, p53, promovendo o crescimento e sobrevivência celular [ 70 , 71 ]. A LMP-1 ativa a via Janus kinase 3 (JAK3)/Signal transducer and ativador of transcription 3 (STAT3), que aumenta a expressão do fator de crescimento endotelial vascular (VEGF) e promove metástase tumoral e invasividade [ 72 ]. LMP-1 também promove a proliferação de células infectadas por EBV através da ativação do receptor do fator de crescimento epidérmico (EGFR) e vias de sinalização de AKT/proteína quinase B com ativação a jusante de ciclina E e telomerase, respectivamente [ 73]. Um estudo mais recente mostrou que a ativação das vias AP-1, JAK/STAT e NF-κB pelo CTAR resultou no aumento da expressão do ligante da proteína 1 de morte celular programada (PD-L1), um importante supressor de checkpoint imunológico na imunologia tumoral [ 74 ].
Atividades biológicas da proteína-1 de membrana latente (LMP-1) na tumorigênese. O EBV LMP-1 ativa vias celulares que levam à invasão e metástase do tumor, proliferação celular e inibição da apoptose.
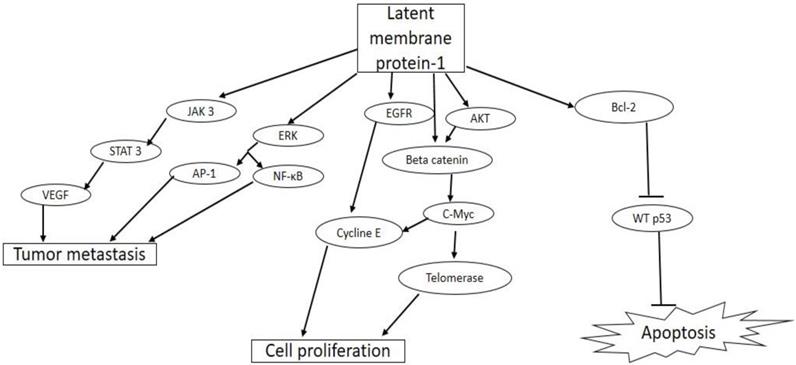
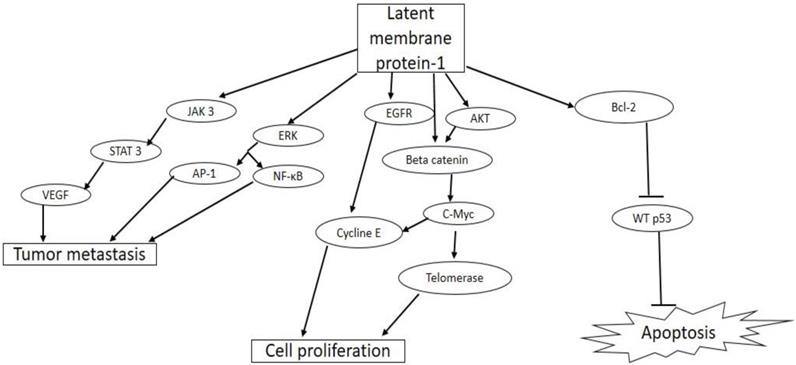{kind=link}
A LMP-2 é composta por duas variantes, LMP-2A e -2B, que são expressas durante a latência II e III em células infectadas com EBV [ 8 ]. Estruturalmente, ambas as proteínas têm 12 domínios transmembranares e um domínio C-terminal citoplasmático de 27 aminoácidos, no entanto, LMP-2A possui um domínio N-terminal citoplasmático adicional de 119 aminoácidos, que não é encontrado em LMP-2B [ 75 ]. O domínio citoplasmático N-terminal de LMP-2A contém o motivo de ativação baseado em tirosina de imunorreceptor (ITAM), que medeia a cascata de sinalização semelhante ao receptor de células B (BCR) em células infectadas com EBV [ 76 , 77 ]. A sinalização semelhante a BCR, mediada por LMP-2A, fornece um forte sinal de sobrevivência que resgata células negativas de BCR da apoptose [ 78 , 79] e inibem a sinalização que leva à reativação lítica [ 80 ]. LMP-2A mostrou ser essencial para a ativação, proliferação e sobrevivência de células B infectadas com EBV em tempos iniciais, após o que é necessário para o crescimento de células B a longo prazo [ 81 ]. Um estudo adicional mostrou que LMP-2A ajuda as células infectadas com EBV a escapar da eliminação por células T CD8 + regulando negativamente (i) a expressão de genes de estágio latente específicos de células T CD8 + , especialmente EBNA-1, (ii) a expressão de MHC proteínas de classe 1 para evitar a apresentação de antígenos e (iii) a expressão do receptor coativador NKG2D para evitar o reconhecimento de células T [ 82 ]. A função do LMP-2B, por outro lado, não é claramente conhecida [ 81], mas um relatório anterior mostrou que interage com LMP-2A para modular as atividades deste último [ 75 ].
EBV-microRNAs (EBV-miRNA)
MicroRNAs (miRNA) são pequenas moléculas de RNA não codificantes, geralmente 18-24 nucleotídeos de comprimento, que regulam negativamente a expressão de mRNAs complementares [ 83 ]. Os miRNAs foram identificados em diferentes organismos, como mamíferos, plantas, algas e vermes, e o EBV foi um dos primeiros vírus identificados a expressar essas pequenas moléculas de RNA não codificantes [ 84 , 85 ]. Verificou-se que o vírus expressa 44 miRNAs em todas as formas de latência e tecidos tumorais, regulando genes celulares e virais .]. Os 44 miRNAs são produzidos a partir de duas regiões de codificação de miRNA: a região BamHI do fragmento H de leitura aberta à direita 1 (BHRF1) que codificou 4 miRNAs, e a região BamHI A de transcrição à direita (BART) que codifica os 40 miRNAs restantes [ 87 ]. Uma vez que os miRNAs do EBV são detectados em tumores associados ao EBV, acredita-se que eles desempenhem papéis importantes na patobiologia do ciclo de vida do EBV e dos tumores associados ao EBV [ 88 ]. Os miRNAs ajudam o vírus a escapar da vigilância imunológica, regulando negativamente a expressão de antígenos virais imunogênicos e proteínas imunes do hospedeiro .]. Por exemplo, Lung e colegas em 2009 relataram que o BART 22, codificado pela região do miRNA BART, regulou negativamente a expressão de LMP-2A, uma proteína viral imunogênica e aumentou a tumorigênese de NPC [ 89 ]. Impotin 7 (IPO7), que é um receptor celular responsável pelo transporte de fatores de transcrição para o núcleo e desempenha um papel fundamental na imunidade inata, demonstrou ser inibido pelo EBV miR-BART3 e miR-BART16 [ 90 , 91 ] e miR-BART2-5p protege as células infectadas por EBV do reconhecimento imunológico por células natural killer (NK) através da inibição da molécula B (MICB) relacionada à cadeia de classe I do complexo principal de histocompatibilidade (MHC) [ 92 , 93]. Outros estudos também demonstraram que o miRNA BART 16 do EBV interrompe a via de sinalização do IFN do tipo I regulando negativamente a expressão da proteína de ligação a CREB, um ativador transcricional do IFN e, portanto, suprimindo a resposta imune do hospedeiro contra o vírus [ 94 ], e o EBV produz BART6-3p que interage com miR 97, um microRNA celular, para suprimir sinergicamente a expressão do receptor de interleucina-6 (IL-6R), que é um receptor para várias citocinas antivirais, como IFN-α, IL-12 e IL-27 [ 95 ], para promover a progressão do tumor em ambos os cânceres de epitélio e linfomas.
Além de direcionar as citocinas celulares, os miRNAs de EBV inibem a destruição de cânceres derivados de células B e células epiteliais por regulação negativa da expressão de genes supressores de tumor; Foi relatado que miR-BHRF1-2 silencia a expressão de PRDM1/Blimp1, que é um supressor tumoral em células B, promovendo, portanto, a linfogênese [ 96 ]. O vírus também tem como alvo a via de sinalização da proteína quinase ativada por mitógeno (MAPK), que suprime a formação de tumor produzindo BART 22 e demonstrou inibir a expressão da MAP quinase quinase quinase 5 (MAP3K5) [ 97 ].
Detecção de EBV
O EBV está implicado em várias malignidades que variam de origens epiteliais e linfoides. Métodos bioquímicos, sorológicos e moleculares para detectar o EBV têm se tornado cada vez mais necessários no diagnóstico e monitoramento de pacientes com doenças associadas ao EBV [ 98 ]. Ensaios sorológicos, que são usados para identificar o vírus em indivíduos infectados, envolvem a detecção de anticorpos contra antígenos específicos do EBV, como antígeno de revestimento viral (VCA), EBNA-1, antígeno precoce (EA) e ácidos nucleicos virais [ 99 - 101 ] . A imuno-histoquímica, que envolve a coloração das principais proteínas de latência do EBV, como LMP-1, LMP-2A, EBNA-1 e -2 em biópsias de tumor, é usada para confirmar a presença do vírus e distinguir entre EBV associado e não EBV -tumores associados [102 ]. O ensaio de hibridização in situ (EISH) de RNA codificado por Epstein Barr (EBER) é considerado o padrão-ouro para a detecção e diagnóstico de infecção por EBV [ 103 ]. A técnica EISH emprega o uso de sondas de ácido nucleico, marcadas ou não marcadas, que podem hibridizar com EBER em seções de parafina de tecidos infectados com EBV [ 104 ]. EBER é um pequeno RNA não codificante e não poliadenilado que é altamente expresso em todas as células infectadas com EBV, independentemente do fenótipo celular. A detecção e quantificação de ácidos nucleicos do EBV em fluidos e tecidos corporais usando a reação em cadeia da polimerase (PCR) também foram exploradas para o diagnóstico e monitoramento de doenças associadas ao EBV [ 105 ].
Doenças causadas por infecção por EBV
O EBV está ligado à patogênese de linfomas, como o linfoma de Burkitt (BL), o linfoma de Hodgkin (HL) e o distúrbio linfoproliferativo pós-transplante (PTLD). As malignidades epiteliais que estão associadas ao EBV nas células epiteliais incluem câncer de nasofaringe (NPC), câncer gástrico (GC) e câncer de mama (BC) [ 34 ]. Em casos raros, foi demonstrado que o EBV infecta células NK e T; causando linfoma de células NK/T do tipo nasal extranodal [ 106 ]. As diferentes malignidades são mostradas na Figura 4. Ao entrar no hospedeiro, o EBV tem tropismo por dois alvos principais, a saber, células B e células epiteliais, bem como alvos não naturais, como células NK/T. O vírus nas várias células-alvo expressa diferentes padrões de genes de latência e estes determinam o tipo de câncer que seria desenvolvido. Os linfomas derivados de células B incluem linfoma de Burkitt, linfoma de Hodgkin e distúrbios linfoproliferativos pós-transplante que são causados pela expressão de latência I, II e III, respectivamente, produtos gênicos. A infecção de células epiteliais pelo EBV resulta em cânceres como câncer de nasofaringe, mama e gástrico, causados pela expressão de latência II, II, I, respectivamente, produtos gênicos. O vírus também pode invadir alvos incomuns, como células natural killer (NK) e linfócitos T, para causar linfoma T/NK do tipo nasal extranodal devido à expressão de produtos do gene de latência II. Detalhes completos de cada tumor são descritos abaixo.
Linfoma de Burkitt (BL)
O linfoma de Burkitt (BL) é uma neoplasia de células B não Hodgkin altamente agressiva e é o câncer pediátrico mais comum do mundo; endêmica na África Subsaariana [ 107 ]. A patogênese do BL tem sido associada à infecção pelo EBV como resultado do isolamento do vírus a partir de linhagens de células BL cultivadas em 1964 [ 5 ]. Desde então, o vírus foi implicado em 95% dos casos de LB de regiões de alto risco e menos de 30% de regiões de baixo risco [ 108 ]. BL ocorre em crianças que vivem em áreas que são holoendêmicas e hiperendêmicas para malária e sugerem que Plasmodium falciparum desempenha um papel na etiologia do câncer [ 109 , 110]. O BL é classificado em três formas diferentes com base em observações clínicas e epidemiologia da doença [ 111 ]. O BL endêmico (eBL) tem uma incidência anual de cerca de 5-10 casos por 100.000 e contribui com 50% de todos os cânceres pediátricos em regiões endêmicas de malária, como a África Equatorial e Papua Nova Guiné, onde o EBV é encontrado em cerca de 95% de todos os casos diagnosticados. casos [ 112 , 113 ]. Os locais comuns de ocorrência de tumores de eBL são os maxilares e o abdome [ 114 ]. O linfoma de Burkitt esporádico (sBL), por outro lado, tem ampla distribuição global, mas tem uma frequência muito menor e é diagnosticado principalmente em crianças e adultos jovens. Nos EUA e na Europa Ocidental, o sBL é responsável por 30-50% de todas as neoplasias pediátricas e menos de 1% em adultos [ 115]. O EBV raramente está relacionado a casos de sBL diagnosticados no mundo ocidental, no entanto, em outros lugares como o Nordeste do Brasil, a frequência de EBV em sBL excede 80% [ 7 , 116 ]. Em contraste, o linfoma de Burkitt associado à imunodeficiência (iBL) foi encontrado em portadores de HIV que desenvolvem o linfoma antes de progredir para AIDS. A incidência de iBL é 10 a 100 vezes maior do que a forma esporádica da doença, e cerca de 30-40% dos iBL são positivos para EBV [ 117 ]. O comprometimento imunológico, que ocorre como resultado da infecção pelo HIV, é responsável pela reativação do EBV de células B latentemente infectadas e, eventualmente, causando rápida progressão para iBL .]. As células T CD4+ iniciam as células T CD8+ contra o EBV circulante em indivíduos imunocompetentes e, assim, colocam o vírus sob vigilância imunológica. No entanto, em indivíduos infectados pelo HIV, a contagem de células T CD4+ é drasticamente reduzida e, portanto, reduz o priming das células T CD8+ específicas do EBV nesses indivíduos [ 119 , 120 ].
A expressão dos genes do EBV em BL é estritamente latente e restrita aos programas padrão de latência tipo I. Apenas EBNA-1 e EBER são expressos em células positivas para EBV em tumores BL [ 121 ]. Embora alguns estudos tenham relatado a detecção de LMP-1 e EBNA-2 em casos raros de eBL e LMP-1 em muitos cânceres de sBL, essas detecções não foram observadas em todas as ocorrências [ 122 - 124 ]. Embora o papel do EBV na patogênese do BL ainda não esteja claro, evidências têm mostrado que a expressão de EBNA-1 em linhagens de células BL promove a proliferação celular inibindo a apoptose [ 125 ]. Relatos recentes sugerem que o EBNA-1 inibe a apoptose em linhagens de células BL, interagindo com proteínas do hospedeiro, como a survivina, uma proteína antiapoptótica e o regulador p53 USP7 [38 , 126 ].
Vista esquemática de epitélios associados ao EBV e malignidades derivadas de células hematopoiéticas.
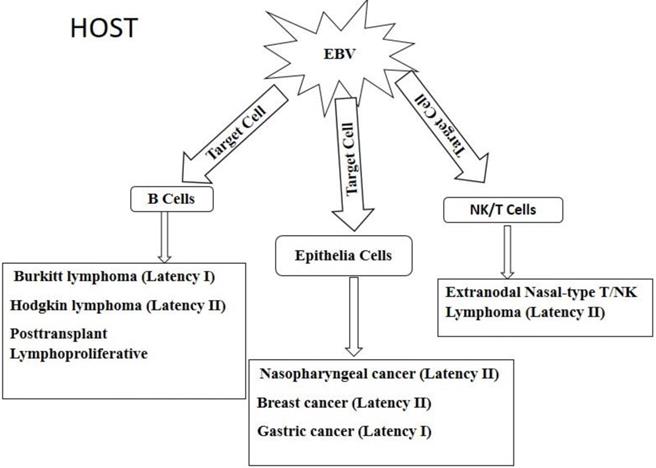
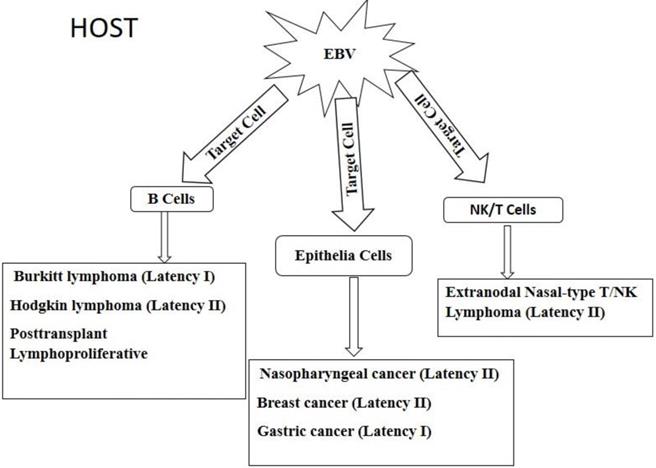{kind=link}
Todos os tumores BL, independentemente de variantes ou status de EBV, são morfologicamente e imunofenotipicamente idênticos e estão relacionados compartilhando padrões gerais de expressão gênica que se assemelham aos dos centroblastos [ 127 , 128 ]. Os BLs, em geral, sofrem translocação cromossômica do proto-oncogene c-myc no cromossomo 8 ou no cromossomo 14 para um dos três loci de imunoglobina, notadamente a imunoglobina de cadeia pesada (Ig) [ 129 ]. Isso traz o c-myc sob o controle do promotor do gene Ig altamente ativo, levando à expressão constitutiva de proteínas c-myc em altos níveis nas células BL. A expressão resulta em crescimento celular descontrolado em BL [ 130 ].
Linfoma de Hodgkin (LH)
O linfoma de Hodgkin (LH) é uma neoplasia linfóide que se origina das células B e se caracteriza pela presença de poucas células gigantes multinucleadas conhecidas como células Hodgkin/Reed Sternberg (HRS), que são circundadas por um infiltrado inflamatório não neoplásico [ 131 ]. A incidência de LH varia de acordo com a distribuição geográfica, sexo, etnia e nível socioeconômico; a incidência é maior nos países desenvolvidos em comparação com os subdesenvolvidos. [ 132 ]. Há, porém, menor mortalidade por LH nos países desenvolvidos em comparação aos menos desenvolvidos; uma observação que tem sido atribuída a melhores instalações de saúde nos países desenvolvidos [ 133]. Cerca de 30-40% dos casos de LH na América do Norte e na Europa foram relatados como positivos para EBV, enquanto em outras partes do mundo, como América Latina, Ásia e África, o EBV foi encontrado em quase 100% de todos os casos de LH [ 134 ] . Apesar da associação do EBV aos casos de LH, o papel do EBV na patogênese desta malignidade permanece desconhecido [ 135 ].
Existem duas variantes de LH com base em diferenças clínicas e morfológicas [ 136 ]. A primeira variante, conhecida como linfoma de Hodgkin predominantemente linfocitário nodular (LLPHL), raramente está associada ao EBV [ 137 ], portanto, não seria discutida nesta revisão. A segunda variante, conhecida como linfoma de Hodgkin clássico (LHC), foi relatada como responsável por cerca de 95% de todos os casos de LH e está principalmente ligada ao EBV [ 138 ]. A associação entre EBV e CHL foi sugerida devido a títulos elevados de anticorpos contra antígenos latentes de EBV, que prosseguiram o desenvolvimento do linfoma após vários anos de infecção .]. A detecção de genes de estágio latente de EBV e RNA de biópsias de pacientes com LHC foi ainda utilizada para confirmar a associação entre a doença e EBV [ 140 ]. A CHL pode ainda ser dividida em quatro subtipos histológicos, a saber, celularidade mista (MC), esclerose nodular (NS), rica em linfócitos (LR) e depleção de linfócitos (LD) [ 138 ]. Cerca de 96% de todos os casos de MC estão associados ao EBV, enquanto a NS está menos frequentemente ligada à infecção por EBV [ 141 ]. Por outro lado, LR e LD raramente estão associados ao EBV [ 142 ].
Doença linfoproliferativa pós-transplante (PTLD)
O distúrbio linfoproliferativo pós-transplante (PTLD) é um tipo de malignidade com complicações com risco de vida em receptores de transplante de órgãos sólidos e aloenxertos de células-tronco hematopoiéticas [ 143 ]. O linfoma ocorre como resultado do aumento da proliferação de células B após o transplante e é principalmente causado pelo EBV [ 144 ]. O genoma do vírus foi encontrado em mais de 90% das células B de pacientes com PTLD, seja após infecção primária, ou reativação de EBV de células infectadas latentemente após tratamento com imunossupressor para evitar a rejeição do aloenxerto [ 145]. O uso de drogas imunossupressoras após o transplante de órgãos reduz o número e a função das células T para influenciar o sucesso do transplante. O uso do imunossupressor leva à depleção de células T específicas do EBV e à ruptura do equilíbrio entre o sistema imunológico e o vírus latente e, consequentemente, há reativação do EBV a partir da latência [ 146 ]. O comprometimento da vigilância imunológica mediada por células T específicas do EBV resulta em blastos linfoproliferativos descontrolados, que eventualmente causam PTLD em receptores de transplante [ 147 , 148 ].
A prevalência de PTLD associada ao EBV varia de 1 a 20%, com incidência variando de acordo com o tipo de aloenxerto, idade e status pré-transplante EBV do receptor do transplante. Os casos de PTLD são mais altos (32%) em receptores de transplante de intestino delgado, e 3-12% de incidência é observada em receptores de transplante de coração, pulmão, fígado e pâncreas. A incidência mais baixa (1-2%) é relatada em receptores de transplante renal [ 146 , 149 ]. Aos 5, 18 e 40 anos, cerca de 50, 20, 5-10% da população geral, respectivamente, não foram expostos ao EBV, portanto, são soronegativos. Esses grupos de pessoas desenvolvem infecção primária após receberem transplante de um doador soropositivo para EBV, predispondo-os a PTLD [ 150 , 151]. A incidência geral de PTLD é maior em crianças do que em adultos devido ao aumento do status soronegativo de EBV pré-transplante entre crianças [ 146 ].
Câncer de nasofaringe (NPC)
O câncer de nasofaringe (CNP) é um tumor maligno do epitélio de células escamosas que se origina da parede lateral da nasofaringe, notadamente, fossa de Rosenmüller e parede posterior superior [ 152 ]. Globalmente, a doença apresenta notável variação na distribuição étnica e geográfica, com cerca de 80% de um total de 65.000 novos casos que são relatados em todo o mundo, ocorrendo no sul da China e no sudeste da Ásia. [ 153 ]. A incidência anual de NPC no sul da China varia entre 20-30 por 100.000 pessoas-ano entre os cantoneses que vivem em Hong Kong e na província de Guangdong, enquanto a incidência em outras partes do mundo, como Europa e Estados Unidos, está abaixo de 1 por 100.000 pessoas/ano [ 154 , 155]. Na África, especificamente no norte da África, a incidência de NPC varia entre 5 a 7 por 100.000 pessoas/ano [ 156 ]. Com base na histologia, a Organização Mundial da Saúde (OMS) classificou o NPC em tipos I, II e III, que são conhecidos como carcinomas de células escamosas queratinizantes, carcinomas não queratinizantes diferenciados e carcinomas não queratinizantes indiferenciados, respectivamente [ 157 ]. Foi proposto um sistema alternativo de classificação mais simples que agrupa NPC em duas variantes histológicas, nomeadamente carcinomas de células escamosas (SCCs) e carcinomas indiferenciados do tipo nasofaríngeo (UCNT) [ 158 ]. Em uma região não endêmica como a América do Norte, cerca de 63% de todos os casos de NPC são UCNT, enquanto no sul da China, cerca de 95% de todos os casos são UCNT [ 159].
O EBV, que é a principal causa de UCNT, foi classificado como agente cancerígeno do grupo 1 pela Agência Internacional para Pesquisa e Câncer (IARC) [ 22 ]. A infecção viral em células epiteliais NPC é de origem clonal; desenvolvendo-se a partir da proliferação clonal de uma única célula epitelial infectada com EBV. Os tumores NPC expressam três produtos gênicos de latência de EBV tipo II (LMP-1, LMP-2, EBNA-1), que são encontrados em cânceres epiteliais implicados em EBV, além da expressão de pequenos RNAs virais codificados, como EBER e microRNAs (miRNAs ) [ 22 , 100 , 160 ]. De todos os produtos do gene de latência do EBV, o LMP-1 foi encontrado em cerca de dois terços dos casos de NPC, indicando o papel fundamental que essa proteína desempenha na tumorigênese.
Como mencionado anteriormente, a infecção das células epiteliais pelo EBV é principalmente lítica e, portanto, os programas líticos padrão são expressos. No entanto, a mudança para programas padrão de latência durante a infecção epitelial por EBV representa um passo fundamental na patogênese da NPC, embora alguns estudos tenham relatado o envolvimento de genes EBV em estágios líticos e latentes na transformação de células epiteliais, o papel dos genes líticos permanece claro [ 15 , 161 , 162 ].
Câncer gástrico associado ao EBV (EBVAGC)
O câncer gástrico (CG) é a terceira principal causa de mortalidade relacionada ao câncer em todo o mundo, com uma incidência anual mundial de mais de 950.000 casos. O câncer gástrico associado ao EBV (EBVAGC) é responsável por cerca de 10% de todos os casos relatados de GC [ 163 , 164 ]. A incidência de EBVaGC mostra variação na distribuição geográfica em todo o mundo, com uma estimativa conjunta na Europa, Ásia e América do Norte e do Sul, sendo 9,2%, 8,3% e 9,9%, respectivamente (Huang et al. , 2014), enquanto que em África, um país como a Zâmbia tem uma frequência de 23% [ 165]. O EBVaGC é classificado em três subtipos histológicos, a saber: carcinoma tipo linfoepitelioma (LELC), tipo adenocarcinoma tipo convencional (CA) e tipo de reação linfóide semelhante à doença de Crohn (CLR) [ 166 ]. O tipo LELC é um carcinoma pouco diferenciado com densa infiltração de linfócitos, semelhante ao NPC. Mais de 80% dos EBVaGCs mostram morfologia do tipo LELC [ 167 ]. O tipo CA, por outro lado, se assemelha morfologicamente a GCs negativos para EBV pela infiltração de linfócitos variáveis com desmoplasia proeminente na ausência de folículos linfóides [ 168 ]. O tipo CLR é caracterizado pela presença de três ou mais folículos linfóides com centros germinativos ativos localizados na borda de avanço da neoplasia [ 168 ].
Uma das características únicas que distingue EBVaGC de não EBVaGC é a proliferação monoclonal de células do epitélio gástrico latentemente infectadas com EBV e sugerindo a presença do vírus nos estágios iniciais da tumorigênese [ 169 ]. Em contraste com GCs EBV-negativos, que ocorrem principalmente no antro como o local patológico predominante, EBVaGC ocorre predominantemente no estômago proximal, incluindo cárdia, fundo e corpo [ 168 ]. Nos estágios iniciais da tumorigênese, o EBVaGC forma uma úlcera nodular bem definida na submucosa com menos fibrose em comparação com os não EBVaGCs, e essa característica patológica é essencial para a ressecção endoscópica da submucosa do tumor [ 170 ].
O genoma do EBV, principalmente os programas padrão de Latência I (EBER, EBNA-1 e BART), foi encontrado nas células do GC e células do epitélio displásico adjacente, mas não pôde ser encontrado em linfócitos circundantes, células estromais, mucosa normal e células intestinais. metaplasia e, portanto, sugere um papel fundamental do EBV na patogênese do GC [ 171 ].
Linfoma de células NK/T do tipo nasal extranodal
Os linfomas NK/T extranodais do tipo nasal são tumores infrequentes que estão regularmente associados à infecção por EBV [ 172 ]. O tumor é caracterizado por extensa angio-invasão e necrose no trato aerodigestivo superior, incluindo cavidade nasal, nasofaringe, seios paranasais e palato [ 173 ]. A doença mostra uma notável variação na distribuição geográfica e por sexo, com a maior incidência ocorrendo no leste da Ásia, México e América do Sul/Central [ 174 ]. O linfoma tem se mostrado predominante entre homens do que mulheres, com proporção de 2-3:1 entre homens e mulheres, e a idade média ao diagnóstico é de 50 anos [ 175 , 176 ].
O EBV expressa programas de latência tipo II (EBNA-1, LMP-1 e -2) nos linfomas extranodais de células NK/T, e o genoma do vírus existe como um epissoma clonal nas células infectadas [ 177 ]. A expressão desses genes demonstrou ativar constitutivamente várias vias de sinalização, incluindo JAK/STAT e NF-κB, para promover o crescimento e a sobrevivência celular [ 178 ]. A expressão de LMP-1 em linfomas extranodais de células NK/T também foi relatada para promover o escape imune tumoral através da regulação positiva do receptor de morte celular programada 1 (PD-1) e do ligante 1 de PD, que são moléculas importantes de checkpoint imunológico em imunologia tumoral [ 179 ].
Análises amplas do genoma relataram anormalidades cromossômicas, notadamente, deleções em 6q21 em linfomas extranodais de células NK/T [ 180 ], o que leva à perda de expressão de muitos genes supressores de tumor, como ATG5, PRDM1, FOXO3, AIM1 e HACE 1 Por exemplo, a perda de FOXO3 e HACE1 em linfomas de células NK/T positivos para EBV resulta em resistência à apoptose através da prevenção da indução de BIM e PUMA e a supressão da ativação de NF-κB induzida por TNF, respectivamente [ 181 ].
Câncer de mama
O câncer de mama é a neoplasia maligna feminina mais frequentemente diagnosticada em todo o mundo e é responsável por cerca de 25% dos casos de câncer incidente entre as mulheres [ 182 ]. Em regiões subdesenvolvidas, como a África Subsaariana, a frequência de câncer de mama é relativamente menor, caracterizada por um curso agressivo e atinge mulheres de idade mais jovem em comparação com o mundo ocidental [ 183 ].
Embora os fatores genéticos, como a herança dos genes 1 e 2 associados ao câncer de mama ( BRCA1/2 ) e o receptor 2 do fator de crescimento epidérmico humano ( HER2 ), além de fatores não genéticos, influenciam o desenvolvimento do câncer de mama, a etiologia permanece desconhecida [ 184 ]. Agentes virais, incluindo EBV, vírus mamário de camundongo (MMV), vírus do papiloma humano (HPV) e citomegalovírus, têm sido implicados em casos de câncer de mama [ 185 - 187 ]. A associação entre EBV e câncer de mama foi relatada em diferentes partes do mundo, incluindo Ásia [ 188 ], África [ 189 ] e Europa [ 190], com prevalência de 30-50%. Subsequentemente, produtos do gene de latência II do EBV, como LMP-1, -2, EBNA- e EBER, foram detectados em células de câncer de mama [ 102 , 191 , 192 ], e um estudo meta-epidemiológico também mostrou que a infecção por EBV foi altamente associado ao risco de desenvolvimento de câncer de mama [ 193 ].
De acordo com a associação entre EBV e câncer de mama, um potencial mecanismo proposto para a transformação de células do epitélio mamário (MEC) sugere que o EBV infecta células que expressam CD21 como receptor de superfície celular, levando à tumorigênese que ocorre através da ativação mediada por LMP-1 de via de sinalização c-MET [ 194 ].
Resumo e estudos futuros
O vírus Epstein Barr é um vírus oncogênico com distribuição global e infectando cerca de 90% da população mundial. Os seres humanos são o único hospedeiro natural do EBV, e o vírus é transmitido pela ingestão de saliva infectada. No hospedeiro, o vírus tem tropismo por duas células principais, células epiteliais e linfócitos B, embora alvos não naturais, como células natural killer e linfócitos T, tenham demonstrado conter o vírus. A presença de EBV em células B tem sido associada a linfomas, que incluem linfoma de Burkitt, linfoma de Hodgkin e distúrbios linfoproliferativos pós-transplante. Por outro lado, a infecção de células epiteliais está implicada na patogénese de malignidades derivadas de células epiteliais tais como cancro nasofaríngeo; Câncer gástrico associado ao EBV e câncer de mama. A patogênese de neoplasias derivadas de células B e de células epiteliais deve-se à expressão de programas de transcrição de EBV ou genes de estágio de latência. Os programas são divididos em latência I (EBNA-1, EBER e BARF0), latência II (EBNA-1, EBER, LMP-1 e -2) e latência III (EBER, EBNA-1,-2,- 3,-4,-5 e LMP-1 e -2A), e sua expressão varia dependendo da diferenciação, tipo e status de ativação da célula alvo. Os programas de latência I estão implicados no linfoma de Burkitt e no câncer gástrico, enquanto a expressão dos programas padrão de latência II está ligada ao câncer de nasofaringe, linfoma de Hodgkin e linfomas de células NK/T. A patogênese dos distúrbios linfoproliferativos pós-transplante tem sido associada à expressão de programas padrão de latência III.
O EBV tem sido associado à patogênese de diferentes malignidades, mas o mecanismo subjacente à patogênese não foi totalmente elucidado. Acredita-se que fatores do hospedeiro e ambientais desempenhem papéis-chave nas malignidades, dificultando o mapeamento claro do mecanismo envolvido na patogênese dos cânceres associados ao EBV. Com o progresso atual na pesquisa genômica, estudos futuros devem se concentrar em como os genes do hospedeiro e os fatores ambientais interagem com os genes virais e como a interação influencia a patogênese das malignidades associadas ao EBV. Devido à sobreposição de sintomas, muitos cânceres implicados pelo EBV são diagnosticados apenas no estágio avançado da doença. Estudos de associação de todo o genoma e sequenciamento do genoma completo de uma grande população de pacientes são, portanto, recomendados para identificar polimorfismo em genes que predispõem os indivíduos aos cânceres associados ao EBV mencionados acima. Os resultados desses estudos ajudarão a identificar populações de alto risco para evolução prognóstica e aumentar as chances de detectar os cânceres em estágios iniciais de desenvolvimento. Estudos futuros devem ser feitos com drogas anticancerígenas que regulam seletivamente a expressão de oncogenes do EBV envolvidos na antiapoptose, proliferação e invasão celular. Finalmente, a inibição seletiva de várias vias de sinalização ativadas por proteínas do EBV oferece uma terapia anticancerígena promissora. Todos os estudos recomendados propostos ajudarão a entender o mecanismo da doença,
Abreviaturas
BC: Câncer de mama; BL: linfoma de Burkitt; EA: Antígeno Precoce; EBER: RNA codificado por Epstein Barr; EBNA: Antígeno nuclear de Epstein Barr; EBV: Vírus Epstein Barr; EBVaGC: câncer gástrico associado a Epstein Barr; GC: Câncer gástrico; HHV-4: Vírus do herpes gama humano 4; LH: linfoma de Hodgkin; IHQ: Imuno-histoquímica; LMP: proteína de membrana latente; NPC: Câncer de nasofaringe; ORF: Quadro de leitura aberto; PBMC: Células mononucleares do sangue periférico; PTLD: Doença linfoproliferativa pós-transplante; VCA: Antígeno de revestimento viral.
Reconhecimentos
Richmond Ayee foi apoiado por um WACCBIP-Banco Mundial ACE M.Phil. bolsa (ACE02-WACCBIP: Awandare) e uma bolsa DELTAS África (DEL-15-007: Awandare). A Iniciativa DELTAS África é um esquema de financiamento independente da Aliança da Academia Africana de Ciências (AAS) para Acelerar a Excelência em Ciência na África (AESA) e apoiada pela Nova Parceria para a Agência de Planejamento e Coordenação do Desenvolvimento da África (Agência NEPAD) com financiamento do Wellcome Trust (107755/Z/15/Z: Awandare) e do governo do Reino Unido. As opiniões expressas nesta publicação são as do(s) autor(es) e não necessariamente as do Banco Mundial, AAS, Agência NEPAD, Wellcome Trust ou o governo do Reino Unido.
Interesses competitivos
Os autores declararam que não existe interesse concorrente.
Referências
1. Schafer G, Blumenthal MJ, Katz AA. Interação de vírus tumorais humanos com receptores de superfície de células hospedeiras e entrada de células . Vírus. 2015; 7 :2592-617
2. Chang CM, Kelly JY, Mbulaiteye SM. et ai . A extensão da diversidade genética do vírus Epstein-Barr e seus padrões geográficos e de doença: uma necessidade de reavaliação . Pesquisa de vírus. 2009; 143 :209-21
3. Longnecker RM, Kieff E, Cohen JI. Vírus de Epstein Barr. Campos Virologia: Sexta Edição: Wolters Kluwer Health Adis (ESP). 2013
4. Sathiyamoorthy K, Hu YX, Möhl BS. et ai . Base estrutural para o tropismo da célula hospedeira do vírus Epstein-Barr mediado pelas glicoproteínas de entrada gp42 e gHgL . Comunicações da natureza. 2016; 7 :13557
5. Epstein MA. Partículas de vírus em linfoblastos cultivados de linfoma de Burkitt . Lanceta. 1964; 1 :702-3
6. Esau D. Causas virais de linfoma: a história do vírus Epstein-Barr e do vírus linfotrópico T humano 1 . Virologia: pesquisa e tratamento. 2017; 8 :1178122X17731772
7. Shannon-Lowe C, Rickinson AB, Bell AI. Linfomas associados ao vírus Epstein-Barr . Phil Trans R Soc B. 2017; 372 :20160271
8. Vírus Rickinson A. Epstein-Barr e sua replicação . Virologia. 2006; 2 :2603-54
9. Saha A, Robertson ES. Linfomas de células B associados ao vírus Epstein-Barr: patogênese e resultados clínicos. Pesquisa Clínica do Câncer. 2011 clincares. 2578.011
10. Thorley-Lawson DA, Hawkins JB, Tracy SI. et ai . A patogênese da infecção persistente pelo vírus Epstein-Barr . Opinião atual em virologia. 2013; 3 :227-32
11. Delecluse H, Bartnizke S, Hammerschmidt W. et ai . Cópias epissômicas e integradas do vírus Epstein-Barr coexistem em linhagens celulares de linfoma de Burkitt . Revista de virologia. 1993; 67 :1292-9
12. Yates J, Warren N, Reisman D. et ai . Um elemento de ação cis do genoma viral de Epstein-Barr que permite a replicação estável de plasmídeos recombinantes em células infectadas latentemente . Anais da Academia Nacional de Ciências. 1984; 81 :3806-10
13. Kang MS, genes latentes do vírus Kieff E. Epstein-Barr . Medicina Experimental e Molecular. 2015; 47 :e131
14. Hatton OL, Harris-Arnold A, Schaffert S. et ai . A interação entre o vírus Epstein-Barr e os linfócitos B: implicações para infecção, imunidade e doença . Pesquisa imunológica. 2014; 58 :268-76
15. Tsao SW, Tsang CM, Para KF. et ai . O papel do vírus Epstein-Barr em malignidades epiteliais . O Jornal de Patologia. 2015; 235 :323-33
16. Santpere G, Darre F, Blanco S. et ai . Análise de todo o genoma de genomas de vírus Epstein-Barr de tipo selvagem derivados de indivíduos saudáveis do Projeto 1000 Genomas . Biologia e evolução do genoma. 2014; 6 :846-60
17. Bouvard V, Baan R, Straif K. et ai . Uma revisão de carcinógenos humanos-Parte B: agentes biológicos . The Lancet Oncologia. 2009; 10 :321
18. Böhm D. Investigação do mecanismo de ativação de NF-kB mediada pela proteína de membrana latente 1 do vírus Epstein-Barr: Freie Universität Berlin, 2010.
19. Corvalán AH, Ruedlinger J, de Mayo T. et al . A Diversidade Filogeográfica do EBV e Ancestralidade Mista nas Américas - Outro Modelo de Coevolução Humano-Patógeno Disrupto . Cânceres. 2019; 11 :217
20. Arvin A, Campadelli-Fiume G, Mocarski E. et ai . Herpesvírus humano: biologia, terapia e imunoprofilaxia. Cambridge University Press. 2007
21. Smatti MK, Al-Sadeq DW, Ali NH. et ai . Epidemiologia do vírus Epstein-Barr, sorologia e variabilidade genética do oncogene LMP-1 entre a população saudável: uma atualização. Fronteiras em Oncologia. 2018:8
22. Banko AV, Lazarevic IB, Folic MM. et ai . Caracterização da variabilidade dos genes do vírus Epstein-Barr em biópsias nasofaríngeas: potenciais preditores para progressão do carcinoma . Plos um. 2016; 11 :e0153498
23. Münz C. Epstein Barr vírus volume 2: um vírus do herpes: muitas doenças. Vol. 391. Springer. 2015
24. Sample J, Young L, Martin B. et ai . Os tipos 1 e 2 do vírus Epstein-Barr diferem em seus genes EBNA-3A, EBNA-3B e EBNA-3C . Revista de virologia. 1990; 64 :4084-92
25. Peh SC, Kim LH, Poppema S. Presença frequente de vírus do subtipo A em malignidades associadas ao vírus Epstein-Barr . Patologia. 2002; 34 :446-50
26. Griffiths P. Citomegalovírus (In: Princípios e Prática de Virologia Clínica. Eds. Zuckerman AJ, Banatvala JE, Pattison JR, Griffiths PD, Schoub BD): John Wiley and Sons, Ltd, West Suusex, Inglaterra. 2004
27. Klemenc P, Marin J, Šoba E. et ai . Distribuição dos genótipos do vírus Epstein-Barr em lavados de garganta, soros, linfócitos do sangue periférico e em biópsias tumorais positivas para EBV de pacientes eslovenos com carcinoma de nasofaringe . Revista de virologia médica. 2006; 78 :1083-90
28. Miller WE, Edwards RH, Walling DM. et ai . Variação de sequência na proteína de membrana latente do vírus Epstein-Barr 1 . Revista de Virologia Geral. 1994; 75 :2729-40
29. da Costa VG, Marques-Silva AC, Moreli ML. A deleção de 30 pb da proteína de membrana latente do vírus Epstein-Barr-1 (LMP1) e polimorfismo XhoI no carcinoma de nasofaringe: uma meta-análise de estudos observacionais . Revisões sistemáticas. 2015; 4:46 _
30. Veja HS, Yap YY, Yip WK. et ai . A deleção de 30 pb da proteína de membrana latente do vírus Epstein-Barr-1 (LMP-1) e a perda de Xho I estão associadas ao carcinoma nasofaríngeo tipo III na Malásia . Revista Mundial de Oncologia Cirúrgica. 2008; 6:18 _
31. AlQarni S, Al-Sheikh Y, Campbell D. et ai . Os linfomas induzidos pelo antígeno nuclear 1 do vírus Epstein-Barr (EBNA1) são dependentes de MDM2. Oncogene. 2018
32. Vírus Kieff E. Epstein-Barr e sua replicação. Virologia de Campos. 2007:2603-54
33. Rowe DT. Imortalização e latência do vírus Epstein-Barr . Frente Biosci. 1999; 4 :D346-D71
34. Thompson MP, vírus Kurzrock R. Epstein-Barr e câncer . Pesquisa Clínica do Câncer. 2004; 10 :803-21
35. Dresang LR, Vereide DT, Sugden B. Identificando sítios ligados pelo antígeno nuclear 1 do vírus Epstein-Barr (EBNA1) no genoma humano: definindo uma matriz de posição ponderada para prever sítios ligados por EBNA1 em genomas virais . Revista de virologia. 2009; 83 :2930-40
36. Amostra J, Henson E, Amostra C. O promotor da proteína nuclear 1 do vírus Epstein-Barr ativo na latência do tipo I é autorregulado . Revista de virologia. 1992; 66 :4654-61
37. Frappier L, Goldsmith K, Bendell L. Estabilização da proteína EBNA1 na origem latente do vírus Epstein-Barr de replicação de DNA por um mecanismo de looping de DNA . Revista de Química Biológica. 1994; 269 :1057-62
38. Dheekollu J, Malecka K, Wiedmer A. et ai . A variante de risco de carcinoma de EBNA1 desregula a latência epissomal do vírus Epstein-Barr . Oncotarget. 2017; 8 :7248
39. Levitskaya J, Coram M, Levitsky V. et ai . Inibição do processamento do antígeno pela região de repetição interna do antígeno nuclear-1 do vírus Epstein-Barr . Natureza. 1995; 375 :685
40. Levitskaya J, Sharipo A, Leonchiks A. et ai . Inibição da degradação proteica dependente de ubiquitina/proteassoma pelo domínio de repetição Gly-Ala do antígeno nuclear do vírus Epstein-Barr 1 . Anais da Academia Nacional de Ciências. 1997; 94 :12616-21
41. Kaiser C, Laux G, Eick D. et ai . O proto-oncogene c-myc é um gene alvo direto do antígeno nuclear do vírus Epstein-Barr 2 . Revista de virologia. 1999; 73 :4481-4
42. Wood CD, Veenstra H, Khasnis S. et ai . Ativação de MYC e silenciamento de BCL2L11 por um vírus tumoral através da reconfiguração em larga escala de hubs potenciadores-promotores . Vida. 2016; 5 :e18270
43. Radkov SA, Bain M, Farrell PJ. et ai . O vírus Epstein-Barr EBNA3C reprime Cp, o principal promotor da expressão de EBNA, mas não tem efeito sobre o promotor do gene celular CD21 . Revista de virologia. 1997; 71 :8552-62
44. Klein G, Ernberg I. Aspectos comparativos de EBV, HHV-8 e vírus tumorais de DNA previamente conhecidos em sistemas experimentais. Herpesvírus humanos. 2007; 29.
45. no IWG de Avaliação. VÍRUS EPSTEIN-BARR . 2012.
46. Wang A, Welch R, Zhao B. et ai . As proteínas do antígeno nuclear 3 do vírus Epstein-Barr (EBNA3) regulam a ligação do EBNA2 a sítios genômicos distintos de RBPJ . Revista de virologia. 2016; 90 :2906-19
47. Saha A, Robertson ES. Impacto da proteína nuclear essencial do EBV EBNA-3C na proliferação e apoptose de células B. Microbiologia do futuro. 2013; 8 :323-52
48. Maruo S, Zhao B, Johannsen E. et ai . Os antígenos nucleares 3C e 3A do vírus Epstein-Barr mantêm o crescimento de células linfoblastóides reprimindo a expressão de p16INK4A e p14ARF . Anais da Academia Nacional de Ciências. 2011; 108 :1919-24
49. Chen A, Zhao B, Kieff E. et ai . Genes celulares regulados por EBNA-3B e EBNA-3C em linhagens de células linfoblastóides imortalizadas pelo vírus Epstein-Barr . Revista de virologia. 2006; 80 :10139-50
50. Alfieri C, Birkenbach M, Kieff E. Eventos precoces na infecção pelo vírus Epstein-Barr de linfócitos B humanos . Virologia. 1991; 181 :595-608
51. Harada S, Kieff E. Epstein-Barr vírus proteína nuclear LP estimula a ativação transcricional mediada pelo domínio ácido EBNA-2 . Revista de virologia. 1997; 71 :6611-8
52. Allan GJ, Inman GJ, Parker BD. et ai . Efeitos do crescimento celular da proteína líder do vírus Epstein-Barr . Revista de virologia geral. 1992; 73 :1547-51
53. Mannick J, Cohen J, Birkenbach M. et ai . A proteína nuclear do vírus Epstein-Barr codificada pelo líder dos RNAs EBNA é importante na transformação de linfócitos B. Revista de virologia. 1991; 65 :6826-37
54. Dufva M, Olsson M, Rymo L. O antígeno nuclear do vírus Epstein-Barr 5 interage com HAX-1, um possível componente da via de sinalização do receptor de células B. Revista de Virologia Geral. 2001; 82 :1581-7
55. Han I, Harada S, Weaver D. et ai . A EBNA-LP associa-se a proteínas celulares, incluindo DNA-PK e HA95 . Revista de virologia. 2001; 75 :2475-81
56. Kashuba E, Yurchenko M, Szirak K. et ai . O EBNA-5 codificado pelo vírus Epstein-Barr liga-se à proteína Fte1/S3a induzida pelo vírus Epstein-Barr . Pesquisa celular experimental. 2005; 303 :47-55
57. Kanamori M, Watanabe S, Honma R. et ai . A proteína líder do antígeno nuclear do vírus Epstein-Barr induz a expressão de quimiocina regulada por timo e ativação em células B. Revista de virologia. 2004; 78 :3984-93
58. Imai T, Nagira M, Takagi S. et ai . Recrutamento seletivo de células Th2 portadoras de CCR4 para células apresentadoras de antígeno pelas quimiocinas CC timo e quimiocina regulada por ativação e quimiocina derivada de macrófagos . Imunologia Internacional. 1999; 11 :81-8
59. Nitsche F, Bell A, Rickinson A. A proteína líder do vírus Epstein-Barr aumenta a transativação mediada por EBNA-2 da expressão da proteína 1 de membrana latente: um papel para o domínio de repetição W1W2 . Revista de virologia. 1997; 71 :6619-28
60. Peng CW, Xue Y, Zhao B. et ai . As interações diretas entre a proteína líder do vírus Epstein-Barr LP e o domínio ácido EBNA2 fundamentam a regulação transcricional coordenada . Anais da Academia Nacional de Ciências. 2004; 101 :1033-8
61. Webster-Cyriaque J, Raab-Traub N. Transcrição de genes do ciclo latente do vírus Epstein-Barr na leucoplasia pilosa oral . Virologia. 1998; 248 :53-65
62. Siegler G, Kremmer E, Gonnella R. et ai . Proteína de membrana latente 1 codificada pelo vírus Epstein-Barr (LMP1) e fatores associados ao receptor de TNF (TRAF): co-localização de LMP1 e TRAF1 na infecção primária por EBV e no linfoma de Hodgkin associado a EBV . Patologia Molecular. 2003; 56 :156
63. Liebowitz D, Wang D, Kieff E. Orientação e patching da proteína de membrana de infecção latente codificada pelo vírus Epstein-Barr . Revista de Virologia. 1986; 58 :233-7
64. Huen D, Henderson S, Croom-Carter D. et ai . A proteína-1 latente de membrana do vírus Epstein-Barr (LMP1) medeia a ativação de NF-kappa B e o fenótipo da superfície celular através de duas regiões efetoras em seu domínio citoplasmático carboxi-terminal . Oncogene. 1995; 10 :549-60
65. Arcipowski KM, Stunz LL, Graham JP. et ai . Mecanismos moleculares da utilização do fator 6 associado ao TNFR (TRAF6) pelo mímico viral oncogênico de CD40, proteína latente de membrana 1 (LMP1) . Revista de Química Biológica. 2011; 286 :9948-55
66. Xie P, Bispo GA. Papéis do fator 3 associado ao receptor de TNF na sinalização para linfócitos B pelas regiões de ativação carboxil-terminal 1 e 2 da proteína de membrana latente da oncoproteína codificada pelo EBV 1 . O Jornal de Imunologia. 2004; 173 :5546-55
67. Lam N, Sugden B. LMP1, um parente viral da família de receptores de TNF, sinaliza principalmente de compartimentos intracelulares . A revista EMBO. 2003; 22 :3027-38
68. Morris MA, Dawson CW, Young LS. Papel da proteína de membrana latente codificada pelo vírus Epstein-Barr-1, LMP1, na patogênese do carcinoma de nasofaringe . Oncologia futura. 2009; 5 :811-25
69. Tsao SW, Tramoutanis G, Dawson CW. et ai . O significado da expressão de LMP1 no carcinoma de nasofaringe. In: Seminários em biologia do câncer . Elsevier. 2002; 12 (6): 473-87
70. Tao Y, Shi Y, Jia J. et ai . Novos papéis e alvos terapêuticos da oncogênese induzida pela proteína 1 de membrana latente codificada pelo vírus Epstein-Barr em carcinoma de nasofaringe. Revisões de especialistas em medicina molecular. 2015:17
71. Hatzivassiliou E, Mosialos G. Vias de sinalização celular envolvidas pela proteína de transformação do vírus Epstein-Barr LMP1 . Frente Biosci. 2002; 7 :d319-d29
72. Wang Z, Luo F, Li L. et ai . A ativação de STAT3 induzida pela proteína de membrana latente do vírus Epstein-Barr1 causa expressão do fator de crescimento endotelial vascular e invasividade celular via sinalização JAK3 e ERK . Revista Europeia de Câncer (Oxford, Inglaterra: 1990). 2010; 46 :2996-3006
73. Chou J, Lin YC, Kim J. et ai . Carcinoma nasofaríngeo-revisão dos mecanismos moleculares da tumorigênese . Pescoço. 2008; 30 :946-63
74. Fang W, Zhang J, Hong S. et ai . LMP1 e IFN-γ acionados por EBV regulam positivamente PD-L1 em carcinoma de nasofaringe: Implicações para terapia onco-alvo . Oncotarget. 2014; 5 :12189-202
75. Rovedo M, Longnecker R. A proteína latente de membrana 2B do vírus Epstein-barr (LMP2B) modula a atividade de LMP2A . Revista de virologia. 2007; 81 :84-94
76. Beaufils P, Choquet D, Mamoun RZ. et ai . O motivo de sinalização (YXXL/I) 2 encontrado nos segmentos citoplasmáticos da proteína do envelope do vírus da leucemia bovina e da proteína 2A da membrana latente do vírus Epstein-Barr pode provocar eventos de ativação precoce e tardia de linfócitos . A revista EMBO. 1993; 12 :5105-12
77. Fruehling S, Longnecker R. O motivo de ativação do imunorreceptor baseado em tirosina do vírus Epstein-Barr LMP2A é essencial para bloquear a transdução de sinal mediada por BCR . Virologia. 1997; 235 :241-51
78. Merchant M, Caldwell RG, Longnecker R. O LMP2A ITAM é essencial para fornecer às células B sinais de desenvolvimento e sobrevivência in vivo . Revista de virologia. 2000; 74 :9115-24
79. Caldwell RG, Wilson JB, Anderson SJ. et ai . O vírus Epstein-Barr LMP2A impulsiona o desenvolvimento e a sobrevivência das células B na ausência de sinais normais do receptor de células B. Imunidade. 1998; 9 :405-11
80. Miller CL, Lee JH, Kieff E. et ai . Uma proteína de membrana integral (LMP2) bloqueia a reativação do vírus Epstein-Barr da latência após a reticulação da imunoglobulina de superfície . Anais da Academia Nacional de Ciências. 1994; 91 :772-6
81. Wasil LR, Tomaszewski MJ, Hoji A. et ai . O efeito da expressão da proteína 2 de membrana latente do vírus Epstein-Barr na cinética da infecção precoce de células B. Plos um. 2013; 8 :e54010
82. Rancan C, Schirrmann L, Hüls C. et ai . A proteína de membrana latente LMP2A prejudica o reconhecimento de células infectadas por EBV pelas células T CD8+ . Patógenos PLoS. 2015; 11 :e1004906
83. Dong M, Chen Jn, Huang Jt. et ai . Os papéis dos microRNAs codificados pelo EBV em tumores associados ao EBV. Revisões críticas em oncologia/hematologia. 2019
84. Alptekin B, Akpinar BA, Budak H. Uma prescrição abrangente para identificação de miRNA de plantas . Fronteiras na ciência das plantas. 2017; 7 :2058
85. Pfeffer S, Zavolan M, Grässer FA. et ai . Identificação de microRNAs codificados por vírus . Ciência. 2004; 304 :734-6
86. Shinozaki-Ushiku A, Kunita A, Isogai M. et ai . Perfil de microRNAs codificados por vírus no carcinoma gástrico associado ao vírus Epstein-Barr e seus papéis na carcinogênese gástrica . Revista de virologia. 2015; 89 :5581-91
87. Barth S, Meister G, Grässer FA. MiRNAs codificados por EBV . Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2011; 1809 :631-40
88. Navari M, Etebari M, Ibrahimi M. et ai . Papéis patobiológicos de microRNAs codificados pelo vírus epstein-barr em linfomas humanos . Revista Internacional de Ciências Moleculares. 2018; 19 :1168
89. Pulmão RW-M, Tong JH-M, Sung YM. et ai . Modulação da expressão de LMP2A por um microRNA codificado pelo vírus Epstein-Barr miR-BART22 recém-identificado . Neoplasia (Nova York, NY). 2009; 11 :1174
90. Yang IV, Wade CM, Kang HM. et ai . Identificação de novos genes que medeiam a imunidade inata usando camundongos endogâmicos . Genética. 2009; 183 :1535-44
91. Dölken L, Malterer G, Erhard F. et ai . Análise sistemática de alvos de microRNA viral e celular em células latentemente infectadas com γ-herpesvírus humanos por ensaio de imunoprecipitação RISC . Hospedeiro celular e micróbio. 2010; 7 :324-34
92. Nachmani D, Stern-Ginossar N, Sarid R. et ai . Diversos microRNAs de herpesvírus têm como alvo o ligante imune MICB induzido por estresse para escapar do reconhecimento pelas células natural killer . Hospedeiro celular e micróbio. 2009; 5 :376-85
93. Bauer S, Groh V, Wu J. et ai . Ativação de células NK e células T por NKG2D, um receptor para MICA induzível por estresse . Ciência. 1999; 285 :727-9
94. Hooykaas MJ, Van Gent M, Soppe JA. et ai . EBV microrna BART16 suprime a sinalização ifn tipo I. O Jornal de Imunologia. 2017; 198 :4062-73
95. Zhang YM, Yu Y, Zhao HP. O EBV-BART-6-3p e o microRNA-197 celular comprometem a defesa imunológica das células hospedeiras no linfoma de Burkitt EBV-positivo . Relatórios de medicina molecular. 2017; 15 :1877-83
96. Ma J, Nie K, Redmond D. et ai . EBV-miR-BHRF1-2 tem como alvo PRDM1/Blimp1: papel potencial na linfomagênese EBV . Leucemia. 2016; 30 :594
97. Chen R, Zhang M, Li Q. et ai . O miR-BART22 codificado pelo vírus Epstein-Barr tem como alvo o MAP3K5 para promover as habilidades proliferativas e invasivas das células hospedeiras no carcinoma nasofaríngeo . Revista do Câncer. 2017; 8 :305
98. Gulley ML. Diagnóstico Molecular de Doenças Relacionadas ao Vírus Epstein-Barr . O Jornal de Diagnóstico Molecular. 2001; 3 :1-10
99. Shao JY, Li YH, Gao HY. et ai . Comparação dos níveis plasmáticos de DNA do vírus Epstein-Barr (EBV) e títulos séricos de anticorpos do antígeno do capsídeo da imunoglobulina A/vírus do EBV em pacientes com carcinoma de nasofaringe . Câncer: Jornal Internacional Interdisciplinar da Sociedade Americana do Câncer. 2004; 100 :1162-70
100. Gao R, Wang L, Liu Q. et ai . Avaliação de sete kits VCA-IgA ELISA recombinantes para o diagnóstico de carcinoma de nasofaringe na China: um estudo caso-controle . BMJ aberto. 2017; 7 :e013211
101. Chen Y, Zhao W, Lin L. et ai . Carga do vírus Epstein-Barr nasofaríngeo: um método complementar eficiente para rastreamento de carcinoma de nasofaringe de base populacional . Plos um. 2015; 10 :e0132669
102. Lorenzetti MA, De Matteo E, Gass H. et ai . Caracterização do padrão de latência do vírus Epstein Barr no carcinoma de mama argentino . PLoS Um. 2010; 5 :e13603
103. Gulley ML, Glaser SL, Craig FE. et ai . Diretrizes para interpretação de hibridização in situ EBER e testes imuno-histoquímicos de LMP1 para detecção do vírus Epstein-Barr no linfoma de Hodgkin . Jornal americano de patologia clínica. 2002; 117 :259-67
104. Cao P, Zhang M, Wang W. et ai . A hibridização in situ de fluorescência é superior para monitorar a carga viral de Epstein Barr em pacientes com mononucleose infecciosa . BMC doenças infecciosas. 2017; 17 :323
105. Gulley ML, Tang W. Usando ensaios de carga viral de Epstein-Barr para diagnosticar, monitorar e prevenir transtorno linfoproliferativo pós-transplante . Clin Microbiol Rev. 2010; 23 :350-66
106. Fox CP, Shannon-Lowe C, Rowe M. Decifrando o papel do vírus Epstein-Barr na patogênese das linfoproliferações de células T e NK . Herpesviridae. 2011; 2 :8
107. Rochford R, Moormann AM. Linfoma de Burkitt. Vírus Epstein Barr Volume 1: Springer. 2015:267-85
108. Magrath IT. Linfoma de Burkitt Africano. História, biologia, características clínicas e tratamento . O jornal americano de hematologia/oncologia pediátrica. 1991; 13 :222-46
109. God JM, Haque A. Burkitt linfoma: patogênese e evasão imune. Revista de oncologia 2010. 2010
110. Thorley-Lawson D, Deitsch KW, Duca KA. et ai . A ligação entre a malária por Plasmodium falciparum e o linfoma de Burkitt endêmico – uma nova visão de um enigma de 50 anos . Patógenos PLoS. 2016; 12 :e1005331
111. Swerdlow SH, Campo E, Pileri SA. et ai . A revisão de 2016 da classificação da Organização Mundial da Saúde (OMS) de neoplasias linfóides . Sangue. 2016; 27 (20):2375-90
112. Bornkamm GW. Vírus Epstein-Barr e a patogênese do linfoma de Burkitt: mais perguntas do que respostas . Revista Internacional de Câncer. 2009; 124 :1745-55
113. Lenze D, Leoncini L, Hummel M. et ai . Os diferentes subtipos epidemiológicos do linfoma de Burkitt compartilham um perfil homogêneo de micro RNA distinto do linfoma difuso de grandes células B. Leucemia. 2011; 25 :1869
114. Aguilar R, Casabonne D, O'Callaghan-Gordo C. et ai . Avaliação do efeito combinado de infecções pelo vírus Epstein-Barr e Plasmodium falciparum no linfoma de Burkitt endêmico usando uma abordagem sorológica Multiplex . Fronteiras em imunologia. 2017; 8 :1284
115. Blum KA, Lozanski G, Byrd JC. Leucemia e linfoma de Burkitt adulto . Sangue. 2004; 104 :3009-20
116. Queiroga EM, Gualco G, Weiss LM. et ai . O linfoma de Burkitt no Brasil é caracterizado por características clínico-patológicas geograficamente distintas . Jornal americano de patologia clínica. 2008; 130 :946-56
117. Gloghini A, Dolcetti R, Carbone A. Linfomas que ocorrem especificamente em pacientes infectados pelo HIV: da patogênese à patologia. In: Seminários em biologia do câncer . Elsevier. 2013; 23 (6): 457-67
118. Amria S, Cameron C, Stuart R. et ai . Defeitos na apresentação do antígeno HLA classe II em linfomas de células B. Leucemia e linfoma. 2008; 49 :353-5
119. Pietersma F, Piriou E, van Baarle D. Vigilância imunológica de células B infectadas por EBV e o desenvolvimento de linfomas não-Hodgkin em pacientes imunocomprometidos . Leucemia e linfoma. 2008; 49 :1028-41
120. Sebelin-Wulf K, Nguyen TD, Oertel S. et ai . Análise quantitativa dos números de células T CD4/CD8 específicas de EBV, números absolutos de células T CD4/CD8 e carga de EBV em receptores de transplante de órgãos sólidos com PLTD . Imunologia de transplante. 2007; 17 :203-10
121. Tsao SW, Tsang CM, Lo KW. Infecção pelo vírus Epstein-Barr e carcinoma de nasofaringe . Phil Trans R Soc B. 2017; 372 :20160270
122. Kelly GL, Milner AE, Baldwin GS. et ai . Três formas restritas de latência do vírus Epstein-Barr neutralizando a apoptose em células de linfoma de Burkitt que expressam c-myc . Anais da Academia Nacional de Ciências. 2006; 103 :14935-40
123. Niedobitek G, Agathangelou A, Rowe M. et ai . Expressão heterogênea de proteínas latentes do vírus Epstein-Barr no linfoma de Burkitt endêmico . Sangue. 1995; 86 :659-65
124. Carbone A, Gloghini A, Zagonel V. et ai . Expressão da proteína de membrana latente 1 codificada pelo vírus Epstein-Barr em linfomas de Burkitt não endêmicos . Sangue. 1996; 87 :1202-4
125. Kirchmaier AL, Sugden B. Dominante-negativos inibidores de EBNA-1 de vírus Epstein-Barr . Revista de virologia. 1997; 71 :1766-75
126. Frappier L. Contribuições do antígeno nuclear de Epstein-Barr 1 (EBNA1) para a imortalização e sobrevivência da célula . Vírus. 2012; 4 :1537-47
127. Hummel M, Bentink S, Berger H. et ai . Uma definição biológica do linfoma de Burkitt a partir de perfis transcricionais e genômicos . New England Journal of Medicine. 2006; 354 :2419-30
128. Dave SS, Fu K, Wright GW. et ai . Diagnóstico molecular do linfoma de Burkitt . New England Journal of Medicine. 2006; 354 :2431-42
129. Guikema JE, Schuuring E, Kluin PM. Estrutura e consequências dos pontos de interrupção do switch IGH no linfoma de Burkitt . Jornal das Monografias do Instituto Nacional do Câncer. 2008; 2008 :32-6
130. O dia todo MJ. Como o vírus Epstein-Barr (EBV) complementa a ativação do Myc na patogênese do linfoma de Burkitt? . In: Seminários em Biologia do Câncer. 2009; 19 (6): 366-76
131. Mirsadraee S, Farzadnia M, Heidary F. et ai . Diagnóstico Imunohistoquímico Comparativo do Linfoma de Hodgkin pelo Método Convencional . revista médica da universidade de ciências médicas de mashhad. 2010; 53 :214-21
132. Mozaheb Z, Aledavood A, Farzad F. Distribuições dos principais subtipos de malignidades linfóides entre adultos em Mashhad, Irã . Epidemiologia do câncer. 2011; 35 :26-9
133. Salati M, Cesaretti M, Macchia M. et ai . Panorama epidemiológico do linfoma de Hodgkin na bacia do Mediterrâneo. Revista mediterrânea de hematologia e doenças infecciosas. 2014:6
134. Glaser SL, Lin RJ, Stewart SL. et ai . Doença de Hodgkin associada ao vírus Epstein-Barr: características epidemiológicas em dados internacionais . Revista Internacional de Câncer. 1997; 70 :375-82
135. Massini G, Siemer D, Hohaus S. EBV em linfoma de Hodgkin. Revista mediterrânea de hematologia e doenças infecciosas. 2009:1
136. Vockerodt M, Cader FZ, Shannon-Lowe C. et ai . Vírus Epstein-Barr e a origem do linfoma de Hodgkin . Revista chinesa de câncer. 2014; 33 :591
137. Wang S, Medeiros LJ, Xu-Monette ZY. et ai . Linfoma de Hodgkin predominantemente linfocitário nodular positivo para vírus Epstein-Barr . Anais de patologia diagnóstica. 2014; 18 :203-9
138. Swerdlow SH. Classificação da OMS de tumores de tecidos hematopoiéticos e linfoides . Classificação de tumores da OMS. 2008; 22008 :439
139. Levine PH, Ablashi DV, Berard CW. et ai . Títulos elevados de anticorpos para o vírus Epstein-Barr na doença de Hodgkin . Câncer. 1971; 27 :416-21
140. Wu TC, Mann RB, Charache P. et ai . Detecção da expressão do gene EBV em células de Reed-Sternberg da doença de Hodgkin . Revista Internacional de Câncer. 1990; 46 :801-4
141. Pallesen G, Hamilton-Dutoit SJ, Rowe M. et ai . Expressão de proteínas replicativas do vírus Epstein-Barr em células de linfoma não Hodgkin relacionadas à AIDS . O Jornal de Patologia. 1991; 165 :289-99
142. Čičkušić E, Mustedanagić-Mujanović J, Iljazović E. et ai . Associação de linfoma de Hodgkin com infecção pelo vírus Epstein Barr . Revista Bósnia de Ciências Médicas Básicas. 2007; 7:58 _
143. Rasche L, Kapp M, Einsele H. et ai . Doenças linfoproliferativas pós-transplante induzidas por EBV: um desafio persistente em SCT hematopoético alogênico . Transplante de medula óssea. 2014; 49 :163
144. Kim HJ, Ko YH, Kim JE. et ai . Doenças linfoproliferativas associadas ao vírus Epstein-Barr: revisão e atualização na classificação da OMS de 2016 . Jornal de patologia e medicina translacional. 2017; 51 :352
145. Green M. Gestão da doença linfoproliferativa pós-transplante induzida pelo vírus Epstein-Barr em receptores de transplante de órgãos sólidos . American Journal of Transplantation. 2001; 1 :103-8
146. Nijland ML, Kersten MJ, Pals ST. et ai . Doença Linfoproliferativa Pós-Transplante Positivo para o Vírus Epstein-Barr Após Transplante de Órgãos Sólidos: Patogênese, Manifestações Clínicas, Diagnóstico e Manejo. Transplante direto. 2016:2
147. Stojanova J, Caillard S, Rousseau A. et ai . Doença linfoproliferativa pós-transplante (PTLD): Determinantes farmacológicos, virológicos e outros . Pesquisa farmacológica. 2011; 63 :1-7
148. Neve AL, Martinez OM. Vírus Epstein-Barr: manobras evasivas no desenvolvimento de PTLD . American Journal of Transplantation. 2007; 7 :271-7
149. Allen U, Preiksaitis J, Prática AIDCo. Vírus Epstein-Barr e distúrbio linfoproliferativo pós-transplante em transplante de órgãos sólidos . American Journal of Transplantation. 2013; 13 :107-20
150. Weikert BC, Blumberg EA. Infecção viral após transplante renal: vigilância e manejo . Revista Clínica da Sociedade Americana de Nefrologia. 2008; 3 :S76-S86
151. Macsween KF, Crawford DH. Vírus Epstein-Barr – avanços recentes . The Lancet doenças infecciosas. 2003; 3 :131-40
152. Carle LN, Ko CC, Castelo JT. Carcinoma de nasofaringe . Patologia de cabeça e pescoço. 2012; 6 :364-8
153. Mimi CY, Yuan JM. Epidemiologia do carcinoma de nasofaringe . In: Seminários em Biologia do Câncer. 2002; 12 (6): 421-9
154. Chang ET, Adami HO. A epidemiologia enigmática do carcinoma de nasofaringe . Biomarcadores de Epidemiologia e Prevenção do Câncer. 2006; 15 :1765-77
155. Jia WH, Qin HD. Fatores de risco ambientais não virais para carcinoma de nasofaringe: uma revisão sistemática . In: Seminários em Biologia do Câncer. 2012; 22 (2): 117-26
156. Hila L, Farah F, Ayari H. et ai . Estudo epidemiológico, imuno-histoquímica e estudos de hibridização in situ de carcinomas de nasofaringe: um relato da Tunísia . Patologia Biologia. 2009; 57 :427-9
157. Shanmugaratnam K. tipagem histológica de carcinoma nasofaríngeo. Publicações científicas do IARC. 1978:3-12
158. Micheau C, Rilke F, Pilotti S. Proposta para uma nova classificação histopatológica dos carcinomas da nasofaringe . Jornal Tumor. 1978; 64 :513-8
159. Yoshizaki T, Kondo S, Wakisaka N. et ai . Papel patogênico da proteína de membrana latente do vírus Epstein-Barr-1 no desenvolvimento de carcinoma de nasofaringe . Cartas de câncer. 2013; 337 :1-7
160. Wu TC, Mann RB, Epstein JI. et ai . Expressão abundante de RNA nuclear pequeno EBER1 em carcinoma de nasofaringe. Um alvo morfologicamente distinto para detecção do vírus Epstein-Barr em espécimes de carcinoma fixados em formalina e incluídos em parafina . A revista americana de patologia. 1991; 138 :1461
161. Lee CP, Chen JY, Wang JT. et ai . A quinase BGLF4 do vírus Epstein-Barr induz a condensação cromossômica prematura através da ativação da condensina e da topoisomerase II . Revista de virologia. 2007; 81 :5166-80
162. Ma SD, Yu X, Mertz JE. et ai . Um mutante do vírus Epstein-Barr (EBV) com expressão aumentada de BZLF1 causa linfomas com infecção abortiva por EBV lítico em um modelo de camundongo humanizado. Revista de virologia. 2012 JV. 00770-12
163. Shibata D, adenocarcinoma gástrico associado ao vírus Weiss L. Epstein-Barr . A revista americana de patologia. 1992; 140 :769
164. Jácome AAdA, Lima EMd, Kazzi AI. et ai . Câncer gástrico positivo para vírus Epstein-Barr: um subtipo molecular distinto da doença? . Revista da Sociedade Brasileira de Medicina Tropical. 2016; 49 :150-7
165. Naseem M, Barzi A, Brezden-Masley C. et ai . Perspectivas sobre o câncer gástrico associado ao vírus Epstein-Barr . Revisões do tratamento do câncer. 2018; 66 :15-22
166. Shinozaki-Ushiku A, Kunita A, Isogai M. et ai . Perfil de microRNAs codificados por vírus no carcinoma gástrico associado ao vírus Epstein-Barr e seus papéis na carcinogênese gástrica. Revista de virologia. 2015 JV. 03639-14
167. van Beek J, Zur Hausen A, Klein Kranenbarg E. et ai . Adenocarcinomas gástricos EBV-positivos: uma entidade clínico-patológica distinta com baixa frequência de envolvimento de linfonodos . Revista de Oncologia Clínica. 2004; 22 :664-70
168. Song HJ, Kim KM. Patologia do carcinoma gástrico associado ao vírus epstein-barr e sua relação com o prognóstico . Intestino e fígado. 2011; 5 :143
169. Uozaki H, vírus Fukayama M. Epstein-Barr e carcinoma gástrico-carcinogênese viral através de mecanismos epigenéticos . Revista Internacional de Patologia Clínica e Experimental. 2008; 1 :198
170. Lee JY, Kim KM, Min BH. et ai . Carcinomas gástricos precoces semelhantes ao linfoepitelioma associado ao vírus Epstein-Barr e dissecção endoscópica da submucosa: série de casos . Jornal Mundial de Gastroenterologia: WJG. 2014; 20 :1365
171. Shinozaki-Ushiku A, Kunita A, Fukayama M. Atualização sobre vírus Epstein-Barr e câncer gástrico . Revista Internacional de Oncologia. 2015; 46 :1421-34
172. Neparidze N, Lacy J. Malignidades associadas ao vírus epstein-barr: patobiologia, características clínicas e tratamentos em evolução . Clin Adv Hematol Oncol. 2014; 12 :358-71
173. Chan J. Linfoma extranodal de células NK/T, tipo nasal. Classificação da OMS de tumores de tecidos hematopoiéticos e linfoides. 2008:285-8
174. Shannon-Lowe C, Rickinson AB, Bell AI. Linfomas associados ao vírus Epstein-Barr . Transações filosóficas da Royal Society B: Ciências Biológicas. 2017; 372 :20160271
175. Au Wy, Weisenburger DD, Intragumtornchai T. et ai . Diferenças clínicas entre linfoma natural killer/célula T nasal e extranasal: um estudo de 136 casos do International Peripheral T-Cell Lymphoma Project . Sangue. 2009; 113 :3931-7
176. Chim C, Ma E, Loong F. et ai . Dicas de diagnóstico para linfoma de células natural killer: apresentação nodal primária e o papel da hibridização in situ para RNA pequeno precoce codificado pelo vírus Epstein-Barr na detecção de envolvimento oculto da medula óssea . Revista de patologia clínica. 2005; 58 :443-5
177. Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-Linfoproliferações Positivas de Derivação de Células BT e NK em Hospedeiros Não Imunocomprometidos . Patógenos. 2018; 7:28 _
178. Jiang L, Gu ZH, Yan ZX. et ai . O sequenciamento do exoma identifica mutações somáticas de DDX3X em linfoma natural killer/células T. Genética da natureza. 2015; 47 :1061
179. Bi Xw, Wang H, Zhang Ww. et ai . PD-L1 é regulado positivamente por LMP1 dirigido por EBV através da via NF-κB e correlaciona-se com mau prognóstico em linfoma natural killer/células T. Revista de hematologia e oncologia. 2016; 9 :109
180. Nakashima Y, Tagawa H, Suzuki R. et ai . Hibridação genômica comparativa baseada em ampla matriz de genoma de linfoma/leucemia de células natural killer: diferentes padrões de alteração genômica de leucemia de células NK agressiva e linfoma de células Nk/T extranodal, tipo nasal . Genes, Cromossomos e Câncer. 2005; 44 :247-55
181. Huang Y, de Leval L, Gaulard P. Molecular underpinning of extranodal NK/T-cell linfoma . Melhor prática e pesquisa Hematologia clínica. 2013; 26 :57-74
182. Shadmani FK, Mansori K, Khazaei S. et ai . Distribuição geográfica da incidência de câncer de mama no Irã . Pesquisa e Terapia Biomédica. 2017; 4 :1295-304
183. Fregene A, Newman LA. Câncer de mama na África Subsaariana: como se relaciona com o câncer de mama em mulheres afro-americanas? . Câncer: Jornal Internacional Interdisciplinar da Sociedade Americana do Câncer. 2005; 103 :1540-50
184. Sun YS, Zhao Z, Yang ZN. et ai . Fatores de risco e prevenções do câncer de mama . Revista Internacional de Ciências Biológicas. 2017; 13 :1387
185. Salman NA, Davies G, Majidy F. et ai . Associação de Papilomavírus Humano de Alto Risco e Câncer de Mama: Um Estudo Baseado no Reino Unido. Rep. Científico 2017:7
186. Pasquereau S, Al Moussawi F, Karam W. et ai . Citomegalovírus, Macrófagos e Câncer de Mama . O Jornal Aberto de Virologia. 2017; 11 :15-27
187. Glenn WK, Heng B, Delprado W. et ai . Vírus Epstein-Barr, papilomavírus humano e vírus do tumor mamário de camundongo como vírus múltiplos no câncer de mama . Plos um. 2012; 7 :e48788
188. Joshi D, Quadri M, Gangane N. et ai . Associação da infecção pelo vírus Epstein Barr (EBV) com câncer de mama em mulheres rurais indianas . PLoS Um. 2009; 4 :e8180
189. Hachana M, Amara K, Ziadi S. et ai . Investigação do vírus Epstein-Barr em carcinomas de mama na Tunísia . Patologia-Pesquisa e Prática. 2011; 207 :695-700
190. Marrão G, Habib M, Paiva A. et ai . A infecção pelo vírus Epstein-Barr e o resultado clínico em pacientes com câncer de mama se correlacionam com a resposta das células imunes TNF-α/IFN-γ . Câncer BMC. 2014; 14 :665
191. Arbach H, Viglasky V, Lefeu F. et ai . Genoma e Expressão do Vírus Epstein-Barr (EBV) em Tecidos de Câncer de Mama: Efeito da Infecção por EBV de Células de Câncer de Mama na Resistência ao Paclitaxel (Taxol) . J Virol. 2006; 80 :845-53
192. Fessahaye G, Elhassan AM, Elamin EM. et ai . Associação do vírus Epstein - Barr e câncer de mama na Eritreia. Agentes Infecciosos e Câncer. 2017:12
193. Bae JM, Kim EH. Infecção pelo vírus Epstein-Barr e risco de câncer de mama: uma meta-análise adaptativa para estudos de caso-controle. Arquivos de Doenças Infecciosas Clínicas. 2016:11
194. Hu H, Luo ML, Desmedt C. et ai . A infecção pelo vírus Epstein-Barr de células epiteliais mamárias promove transformação maligna . EBioMedicina. 2016; 9 :148-60
Contato do autor
![]() Autor correspondente: Osbourne Quaye, Departamento de Bioquímica, Biologia Celular e Molecular, Universidade de Gana, Volta Road, PO Box LG 54, Legon, Accra-Gana; Telefone: +233-2611-70904.
Autor correspondente: Osbourne Quaye, Departamento de Bioquímica, Biologia Celular e Molecular, Universidade de Gana, Volta Road, PO Box LG 54, Legon, Accra-Gana; Telefone: +233-2611-70904.
Recebido 2019-6-3
Aceito 2019-12-1
Publicado 2020-1-17
Comentários
Postar um comentário